炔烃区域选择性氢化反应的脱羧方法外文翻译资料
2023-01-05 14:12:49
炔烃区域选择性氢化反应的脱羧方法
摘要:芳烃C-H键的区域选择性活化是芳烃官能化反应的长期挑战,例如炔烃的氢化反应。一种可能的解决方案是使用可移除的导向基团,其激活几种芳族C-H键中的一种。在这里,我们报告了一种新的用苯甲酸衍生物进行区域选择性炔烃氢化反应的催化方法,其中羧酸官能团将炔烃引导至邻-C-H键并原位消除以形成乙烯基芳烃产物。该串联序列的脱羧阶段设想在邻烯基部分的协助下进行,该烯基部分由最初的炔烃偶联形成。这种钌催化的脱羧炔烃氢化反应消除了对苯甲酸预先存在的用于底物活化的邻位取代,在氧化还原中性和相对温和条件下进行的共同需要,并且耐受广泛的合成有用的芳族官能团。因此,它显着增加了苯甲酸作为易接近的芳香族构件的合成效用。
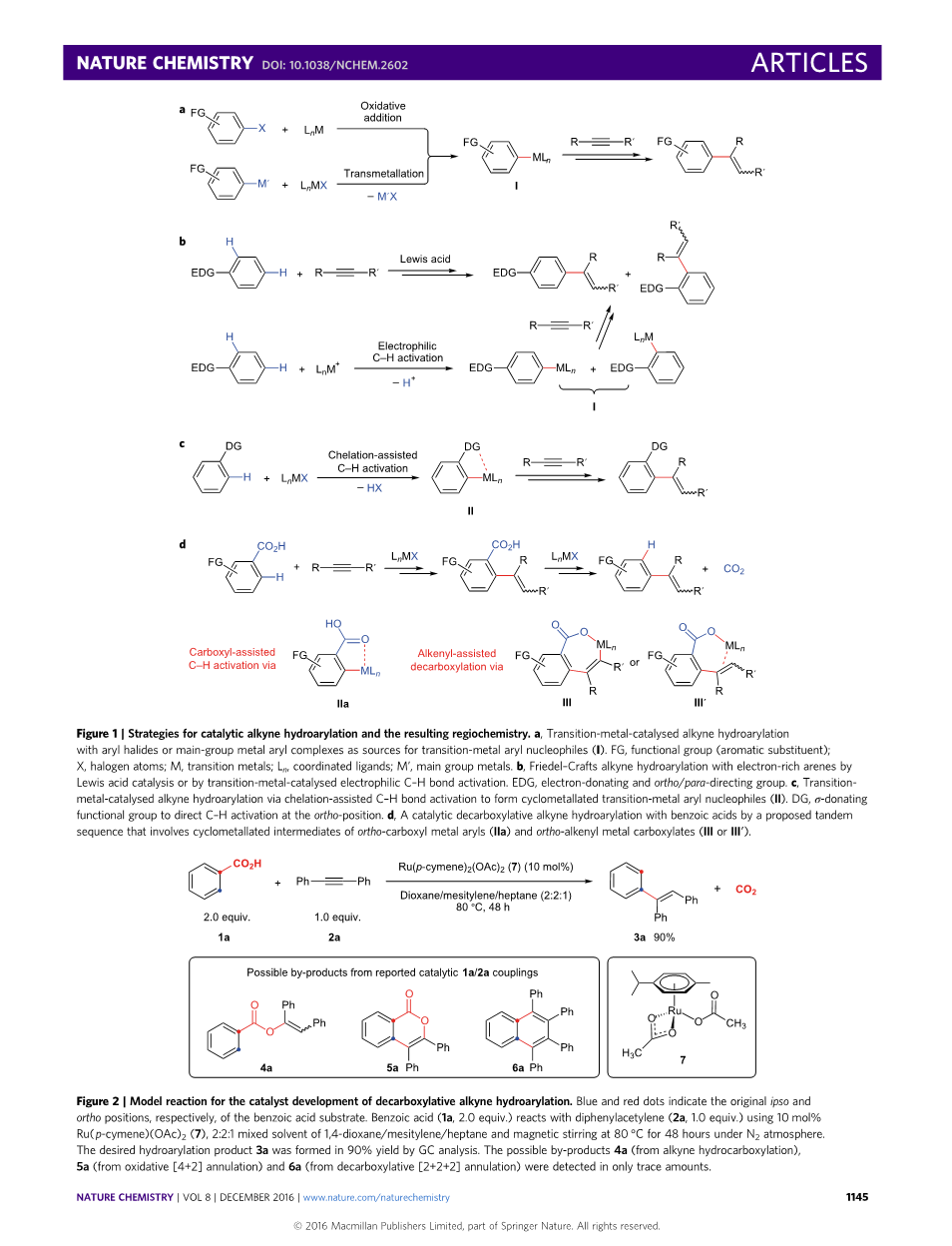
芳基取代的烯烃是生物活性化合物中的正价结构和用于精细化学品和材料的重要合成中间体。由于两种试剂的商业和合成有效性,向炔烃添加芳烃是这些结构的一种有吸引力的途径。炔烃芳基C-H键活化反应的原子效率更高,比炔烃与预先官能化的芳香化合物的催化偶联反应生成更少的盐类废产物(图1a)。然而,实际炔烃芳基C-H键活化反应长期面临的挑战是控制多个芳烃C-H键之一的反应的区域选择性。 例如,经过Friedel-Crafts过程的炔烃芳基C-H键活化反应需要给电子芳烃取代基,以获得令人满意的反应性(图1b),并且这些取代基的导向作用,通常导致邻-和对-烯基芳烃异构体的混合物。
近年来,已经开发了各种过渡金属催化剂用于炔烃芳基C-H键活化反应,通过芳基C-H键的形式活化形成金属-芳基中间体。许多报道的这些催化剂都是Lewis酸性的,并且通过亲电芳香取代裂解了C-H键,这在机理上类似于Friedel-Crafts反应以及在区域选择性上有类似的局限性(图1b)。另一种方法是通过环金属化在配位官能团的邻位直接断裂C-H键(图1c),这在Lewis和Smith关于催化邻位的环金属化钌(II)配合物的报道中首次证明苯酚与乙烯的烷基化。在螯合辅助的C-H官能化策略的基础上,Murai及其同事进一步探索了钌催化下烯烃和炔烃与芳香酮的氢化反应。自这些开创性研究以来,螯合辅助炔烃氢化反应已探索各种过渡金属催化剂以及宽范围的基于杂原子的导向基团。然而,从芳基C-H键活化产物中除去这些邻位引导基需要另外的化学转化,并不能总是实现。因此,通过炔烃芳基C-H键活化以获得邻位取代的链烯基芳烃受到邻位定向基团官能团性质的限制,并且炔烃芳基C-H活化不能通过现有方法选择性地获得间位或对位取代的链烯基芳烃。
我们假设用芳基羧酸反应可以解决控制炔烃氢化芳基化的区域选择性的问题。羧基官能团可作为邻位定向基团,在C-H烯基化后通过有金属中间体的脱羧反应将其除去,得到具有乙烯基间位或对位残基取代基的产物(图1d)。炔烃氢化反应的脱羧反应将使用简单易得的苯甲酸作为易接近的芳族结构单元,CO2作为唯一的副产物并且不含盐废物。但苯甲酸衍生物的催化脱羧反应通常较慢,并且需要邻位取代的sigma;-吸电子基或空间位阻基团促进底物活化。绝大多数此类脱羧转化也需要高反应温度,通常高于140℃,因此明显地限制了底物范围。为了获得较高反应活性,许多脱羧方法还利用化学计量银(I)或铜(II)盐,其与易氧化官能团的兼容性较差,尽管Goobeta;en和其合作者已明显减少盐副产物的量。
我们认为氢化芳基化反应可以绕过这些问题,因为在C-C键形成后,连结上关键中间体的烯烃单体可以用作脱羧的活化单元,并且在苯甲酸反应物中不需要这样的基团。在一系列论文的一章或在单独的步骤中已经发表过将羧基官能团可作为能移除的邻位定向基团,但未发表与炔烃的C-H键脱羧烯基化。报道的绝大多数C-H脱羧功能化过程需要高反应温度(130-170℃)以及化学计量的Ag(I)或Cu(II)盐来促进脱羧。另外,大多数进行这些反应的苯甲酸须含有用于活化的邻位取代基,只有少数仍需超过120℃的高反应温度(参考文献35,40,46,47)。因此,我们探求区域选择性炔烃氢化方法的新策略,为在温和条件下羧基导向的且具有位点选择性的原位消除反应。
我们发现了一种新的的催化剂,可用于不同苯甲酸的脱羧炔烃氢化反应,其克服了脱羧范围的局限性,可用大量邻位、间位和对位芳基取代基合成得到烯基芳烃,并在足够温和的条件下合成一系列有用的官能团(图1d)。该催化过程通过包含初始C-H活化阶段的串联序列进行操作,在该阶段中羧基引起特定的C-H键添加至金属并且炔随后插入金属-芳基键中以形成邻烯基苯甲酸中间体(IIa)。芳烃与炔烃的偶合之后是脱羧阶段,其中新安装的烯基部分与羧酸酯中间体的金属中心配位以促进CO2释放,或者作为烯基sigma;-供体配体(III)或作为pi;-烯烃配体(III)。由烯基配位产生的低能量脱羧过程有效地消除了先前苯甲酸邻位取代的先决条件,并且在比典型脱羧过程低得多的温度下进行。苯甲酸与各种邻位,间位和对位芳基取代基以及母体苯甲酸有区域选择性,证明了该脱羧炔烃氢化芳基化的底物范围宽。在氧化还原中性及温和的反应条件下,允许与未受保护的易氧化官能团如苯胺和苯酚可兼容。因此,我们的催化剂体系能够使生物质衍生的酚酸发生脱羧炔烃氢化反应,例如香草酸(木质素的降解产物)。
结果与讨论
我们用苯甲酸(图2中的1a)和二苯基乙炔(2a)之间的模型反应开始我们的催化剂开发。8我们重点关注基于钌(II)的催化剂前体,该前体已广泛用于螯合辅助的C-H键官能化,包括炔烃氢化芳基化。钌催化剂很少被用作苯甲酸脱羧促进剂。(4a),氧化[4 2]杂环化(5a)和氧化[2 2 2]杂环化合物反应的多种副产物具有高化学选择性,以促进氢化产物3a。2]通过脱羧(6a)进行碳环化。为此,我们评估了各种反应参数,如钌催化前体配体,盐的加和,溶剂和反应温度。由于苯甲酸及其取代的类似物通常可商购并且价格便宜,因此我们用2当量进行反应。 苯甲酸以最大化炔烃的转化。在本研究中我们监测的所有反应中,炔烃完全转化后过量的苯甲酸未进行反应; 没有观察到苯甲酸的直接原羧酸化。这些实验的选择结果总结在补充表1中。我们发现在80℃时10mol% Ru(p-cymene)(OAc)2(7)促进了1a和2a之间的偶联 C和在2:2:1二噁烷/均三甲苯/庚烷的混合溶剂中。在这些条件下,3a在48小时内 以90%收率选择性地形成;检测到的副产物的总产率小于5%。用混合溶剂系统的单一溶剂进行的反应以类似高水平的高化学选择性3a与混合溶剂一样,但产率较低(补充表1给出了详细说明)。
在确定催化过程的条件下,我们用各种取代的苯甲酸研究了2a的脱羧氢化(表1,条目1-19和29-33)。所有的反应都发生在立体选择性高的芳烃顺式C-H键活化反应。观察到的区域选择性是符合预想串联序列的邻-C-H的烯基化反应和随后的脱羧反应。因此,与对位取代苯甲酸的反应以独特的区域选择性进行,得到间位取代的链烯基芳烃3ba和3c-3p。用强给电子对位取代基(包括甲基,甲氧基和二甲氨基基团(产物3ba,3c和3d))。对于弱给电子的对甲硫基,反应需要100℃的较高温度以产率69%得到产物3e。本方法的氧化还原中性特性不仅保护酰基,而且还保护未保护的苯酚和苯胺官能团以高收率形成间位O-或N-官能化产物3f-3i。相反,含有作为亲电子芳族取代失活基团的对位取代基的芳烃的反应性显着较低。例如,具有对位卤素、乙酰基、甲酰氨基和-CF3基团的苯甲酸即使在100℃的较高反应温度下也导致产物3j-3p产率低或产率一般。类似地,具有对位硝基,甲酰基和甲氧基羰基的苯甲酸导致期望的氢化芳基化产物(3pa-3pc,通过气相色谱(GC)分析小于10%)的低产率和副产物6a的竞争性形成通过[2 2 2]脱羧碳化。
用邻位取代的苯甲酸,应该形成由对位取代的类似物形成的相同的间位取代的烯基芳烃。 这种设想的区域选择性是从含有邻位取代基(包括甲基,甲氧基,羟基,苯基和氟原子)的几种苯甲酸的反应中观察到的(表1,条目29-33)。这些邻位取代的苯甲酸导致间位取代的链烯基芳烃的排他性形成,而没有可检测地形成邻位取代的链烯基芳烃或来自原脱羧基的相应芳烃。这种用邻位取代的苯甲酸进行的原位选择性脱羧转化的缺乏支持了我们提出的涉及与原位连接的邻烯基部分配位的脱羧途径,而不是通过预先存在的邻位取代基的空间性质促进脱羧。
期望间位取代的苯甲酸在羧基的两个不同的邻位上发生竞争性的C-H官能化。 实际上,3-甲氧基苯甲酸与2a反应生成50%总收率和低选择性(产物3r/r(表2))的四个氢化芳基化产物的区域和立体异构体的不可分离的混合物。我们假设间位取代基的电子和空间特性都可能影响区域选择性。更具空间要求的间位取代基应该抑制邻烯基化并促进对位烷基化。 与这一说法相一致,空间要求很高的间二甲氨基,异丙基和叔丁基取代基导致以50-74%的产率独家形成对位烯烃化产物3s-3u。用含有间位和对位取代基的两种原儿茶酸衍生物也观察到芳香族取代基对区域选择性的这种控制。 邻烯基化产物3v的排他性形成提示了主要的电子效应并且由缩醛保护的3,4-二羟基部分产生的空间效应可以忽略不计。相反,与香草酸的反应导致通过区域选择性C-H活化和在甲氧基的对位和羟基的间位的芳族位点的烯基化产生3w的排他性形成。
用4-甲氧基苯甲酸作为反应配体(表1,条目20-28和表2,条目1-5)研究经历该氢化芳基化的炔的范围。从末端炔烃如苯乙炔的反应中未检测到偶联产物;这种产物的不存在可能是由于形成非生产性的Ru(II)炔基或乙烯基茚基配合物。不对称的芳基烷基炔烃与独特的区域选择性反应形成1-烷基-1-间茴香基烯烃产物(3bb-3bf)。甲基,乙基和正丁基取代的苯乙炔比二苯基乙炔(2a)的反应性低并且需要20 mol%的Cu(OAc)2添加剂以60-77%产率得到产物3bb-3bd以及少量的异香豆素副产物5和萘副产物6a来自氧化环化反应。通过添加Cu(OAc)2可以观察到较高的总收率和较低的化学选择性,这可能源自其众所周知的作为钌催化的氧化C-H官能化过程的氧化剂。
我们建议Cu(OAc)2有助于从6a再生活性催化剂。通过将Ru(0)氧化为Ru(II)羧酸盐络合物来再生活性催化剂,其是启动C-H活化所需的,其导致所需的氢化芳基化产物3的形成。与含有简单烷基的炔烃相比,甲氧基甲基取代的芳基乙炔反应更快并且不需要Cu(OAc)2添加剂来实现良好收率(产物3be和3bf)。对于对称的二烷基乙炔(产物3bg-3bi),也观察到甲氧基甲基取代基的反应性增强。与几种对称二芳基乙炔反应的结果提供了关于炔烃反应性(产物3bj-3bo)的空间效应的额外信息。特别是含有邻位取代的芳基的炔烃(产物3bj和3bm)比含有间位或对位取代的芳基的炔烃观察到的产率更高。
我们建议整个催化过程通过一个串联序列发生,涉及邻苯甲酸烯基酯中间体的脱羧反应(图1d)。环状金属化的烯基钌(II)羧酸盐络合物(图3中的III)可能通过羧酸盐引导的C-H活化并随后将炔烃插入Ru-芳基键中形成(补充图1给出了所提出的催化循环的细节)。在III中Ru-烯基键的质子化将产生链烯基螯合的羧酸Ru(II)中间体III(图3中的路径A),其将通过脱羧基形成氢化芳基化产物3,随后质子化所得烯基螯合的Ru(II)芳基中间体IV。另一种脱羧方法是从中间体III直接脱羧,这是Ru-芳基加成穿过CO2中C=O双键的显微镜反向(路径A)。所得到的钌环V经历Ru-芳基的双重质子化和Ru-烯基键以释放氢化芳基化产物3.路径A和路径A都具有通过邻烯基部分的配位辅助的脱羧作用,其通过路径A中的pi;-烯烃络合和路径A中的sigma;-烯基连接而发生。作为竞争过程,从III形成的C-O键还原消除将产生异香豆素5作为氧化[4 2]环化(路径B)的副产物.。
作为III的第三种可能的反应,将第二当量的炔2插入到Ru-烯基键中(路径C)将导致经由连续的脱羧基形成氧化性[2 2 2]副产物6,并且 C-C还原消除闭环。如果链烯基辅助的3m和3be的产率比不存在酸时产生的高(图4a)。其次,2a与Ru(II)苯甲酸Ru(II)配合物Ru(p-cymene)(OBz)2(8)之间的2:1反应在48小时内在室温下导致定量形成Ru(0)络合物 9(图4b)。通过单晶X射线衍射建立了配合物9的固态结构(补充图2和补充表2和3给出了更多细节)。除了eta; 6-对伞花烃配体之外,该络合物还含有eta; 4 -1,2,3,4-四苯基萘配体,并且对于脱羧炔烃氢化芳基化没有显示催化反应性。萘配体大概是由路径C中的[2 2 2]环形产生的。复合物9的化学计量形成的温和温度及其完全缺乏催化活性表明路径C对氢化芳基化过程有害并导致催化剂失活。与该断言相一致,1,2,3,4-四取代的萘(6)为对于脱羧基氢化芳基化反应较弱的缺电子苯甲酸(表1中的产物3l-3p)的反应的主要副产物。然而,更不容易经历两个连续插入反应的更多受阻炔烃以高收率形成了氢化芳基化产物(表1中的产物3bj和3bm)。
我们建议整个催化过程通过一个串联序列发生,涉及邻苯甲酸烯基酯中间体的脱羧反应(图1d)。环状金属化的烯基钌(II)羧酸盐络合物(图3中的III)可能通过羧酸盐引导的C-H活化和随后将炔烃插入到钌-芳基键中形成(补充图1给出了所提出的催化循环的细节)。 在III中Ru-烯基键的质子化将产生链烯基-螯合的羧酸Ru(II)中间体III(图3中的路径A),其将通过脱羧基形成氢化芳基化产物3,随后质子化所得烯基-螯合的 Ru(II)芳基中间体IV。另一种脱羧方法是由中间体III直接脱羧,这是Ru-芳基在CO2中跨C=O双键的微观反向(路径A)。所得到的钌环V经历Ru-芳基和Ru-烯基键的双重质子化以释放氢化芳基化产物3.路径A和路径A两者的特征在于通过邻烯基部分的配位辅助的脱羧,其通过pi;-烯烃络合发生途径A和路径A中的sigma;-烯基连接。作为竞争
剩余内容已隐藏,支付完成后下载完整资料

英语原文共 8 页,剩余内容已隐藏,支付完成后下载完整资料
资料编号:[281153],资料为PDF文档或Word文档,PDF文档可免费转换为Word



