镍基过渡金属碳化物催化剂计算设计与催化产氢机制研究毕业论文
2020-02-13 12:41:30
摘 要
在人类社会快速发展的今天,由于发展所带来的能源安全和环境污染已经成为了人类面临的全球性问题,而催化则被认为是解决这些难题的关键之一。在诸多催化剂中,铂是其中最为优秀的催化剂,然而由于其产量低、价格昂贵等原因,限制了它的发展。如今越来越多的人将发展方向投入到了非贵金属催化剂上。Ni3C作为一种极具潜力的非贵金属催化剂已经在实验上进行了大量研究,为了从理论上解释非贵金属材料Ni3C对于析氢反应(HER)的反应活性,本文采用了第一性原理密度泛函理论分别对Ni3C晶体(113)晶面HER催化的稳定性、几何结构、电子特性以及催化性能进行了综合全面的分析。
论文主要研究了Ni3C晶体(113)两种不同类型的晶面在析氢反应过程中的氢吸附能,过电势以及氢吸附吉布斯自由能,通过与铂进行比较,对其催化性能进行评价。
研究结果表明:富碳的化学计量表面是热力学稳定的,C-Ni3C(113)晶面的桥位在析氢反应中是不可或缺的,因为它不仅显示出很强的电催化活性,而且具有十分合适的氢吸附能、过电位和优异的稳定性。
本文的特色:从理论上系统探究了Ni3C(113)晶面在HER催化方面的机理,通过计算解释了Ni3C(113)晶面的催化性能电子尺度机制。
关键词: Ni3C;催化;析氢反应;
Abstract
Nowadays, with the rapid development of human society, energy security and environmental pollution caused by development have become a global threat to human beings, and catalysis is considered as one of the keys to solve these problems. However, as one of the most excellent catalysts, platinum has limited its development due to its low yield and high price. At present, more and more people devote their development direction to non-precious metal catalysts. Ni3C, as a potential non-noble metal catalyst, has been extensively studied in experiments. In order to examine the reactivity of noble-metal-free Ni3C toward hydrogen evolution reaction (HER) theoretically, we report a comprehensive first-principles density functional theory (DFT) study on the stability, geometric structure, electronic characteristics, and catalytic activity for HER on the Ni3C crystal (113) surfaces。
In this paper, the hydrogen adsorption energy, overpotential and Gibbs free energy of hydrogen adsorption on two different types of Ni3C crystal (113) surfaces during hydrogen evolution reaction were studied. The catalytic performance was evaluated by comparing with platinum.
The results show that the C-rich stoichiometric surface is thermodynamically stable, The bridge-site of C-rich Ni3C (113) is indispensable for HER because it not only displays improved electrocatalytic activity, but also possesses appropriate hydrogen adsorption energy, overpotential and robust stability.
The characteristic of this paper: The mechanism of Ni3C (113) crystal surface in HER catalysis was investigated theoretically, and by calculation, the catalytic performance of Ni3C (113) crystal surface was explained.
Key Words:C-rich Ni3C;electrocatalytic;hydrogen evolution reaction
目 录
第1章 绪论 1
1.1 HER催化 1
1.1.1 HER催化的应用背景 1
1.1.2 HER催化的机理 1
1.1.3 电催化析氢(HER)的主要参数 2
1.2非贵金属析氢催化剂的研究进展 3
1.2.2 金属硫族化物催化剂 4
1.2.3层状过渡金属碳化物和氮化物MXene 5
1.2.4 过渡金属氢氧化物 5
1.3 实验材料介绍 6
1.4 实验意义 6
第二章 实验计算原理 8
2.1 第一性原理 8
2.2密度泛函理论 8
2.2.1 Hohenborg-Kohn定理 8
2.2.2 Kohn-Sham方程 9
2.3 计算软件 9
第3章 实验计算过程 10
3.1 实验参数设置 10
3.2 计算过程 10
第四章 实验结果与分析 12
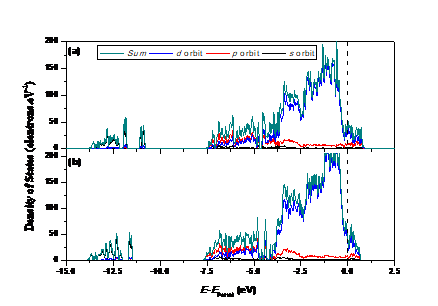
4.1电子结构变化 12
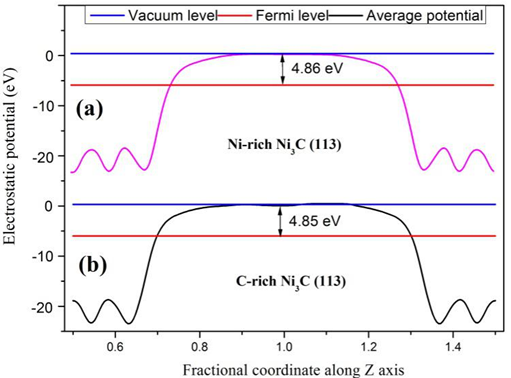
4.2 功函数的计算 13
4.3 氢吸附能以及氢吸附吉布斯自由能的计算 13
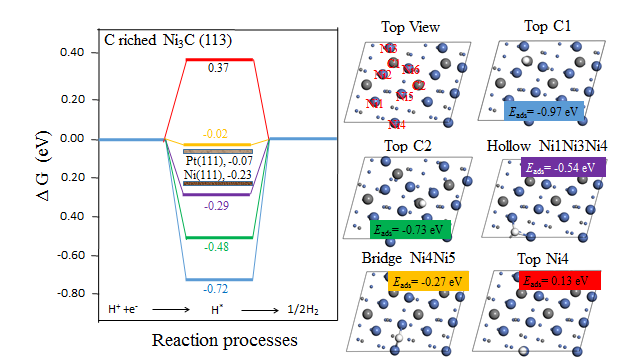
4.3.1 C-rich Ni3C(113)晶面 13
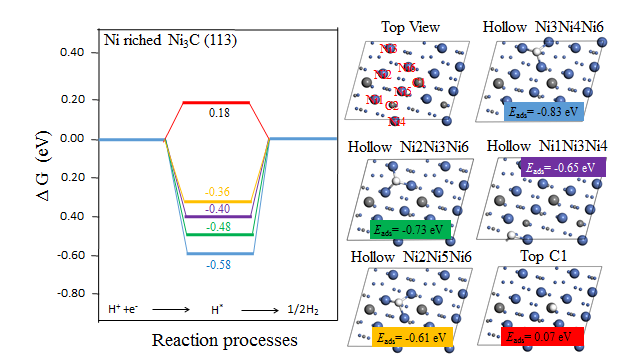
4.3.2 Ni-rich Ni3C(113)晶面 15
4.4 本章小结 16
第五章 总结与展望 18
参考文献 19
致 谢 21
第1章 绪论
1.1 HER催化
1.1.1 HER催化的应用背景
能源是人类社会发展不可或缺的基础,随着社会的快速发展,能源紧缺的问题日益严重。现如今,三大化石燃料(煤、石油以及天然气)依然是世界各国赖以发展的主要能源。在人类快速发展的一百多年里,在大自然界中经历上百万年才形成的化石燃料被大量的开采,不可再生能源的储量急剧减少,能源紧缺以及化石燃料燃烧所造成的环境污染问题逐渐成为了遏制全球发展的重大难题。因此,为了适应人类快速发展的脚步,尝试开发清洁的可持续能源来代替储量日益减少的化石能源已经成为了全球科学研究人员亟待解决的难题。
由于氢气具有较高的热值,而且在燃烧以后生成的是无环境污染的物质水。因此,氢气被认为是一种十分具有发展前景的清洁能源。但是,在大自然界中,氢气并不像化石燃料那样是天然存在的,而是需要人工进行合成。在目前,工业上制取氢气的途径主要包括:甲烷蒸汽重整,煤气化过程和催化反应。显然易见,前两种获取氢气的方法仍需要用到化石燃料,也就是说不能解决空气污染和温室效应等问题。水裂解产氢则为使用经济、合理的途径获取氢气带来了希望。首先,催化产氢的原料是水,水作为地球上储量最为丰富的物质,可以满足大量产氢的需求。其次,催化反应的产物为氢气和氧气,对环境没有任何危害。
在整个催化过程中,包含了两个半反应,即产氢反应(水还原反应)和产氧反应(水氧化反应)。采用合适的催化剂可以有效的降低反应能垒,提高催化的速率。,催化剂的稳定性、成本和效率是限制其商业化应用的几个主要问题。目前来说,铂等贵金属催化剂是最高效的水裂解催化剂,然而,由于这些贵金属产量低,价格高,容易中毒失效等原因,使得该类型催化剂的应用受到了严重的阻碍。所以,对于过渡金属催化剂以及其他高效廉价催化剂的研究成为了科学家们探寻的热点。
1.1.2 HER催化的机理
在本文中,对于Ni3C晶体(113)晶面的催化活性的理论计算主要是基于电催化研究,因此,在这里我们详细介绍一下电催化产氢的机理。首先,电子在电极表面上转移,当催化剂的活性位点上吸附有质子时,电子会与质子结合形成吸附态的氢原子(H*),称为Volmer反应。之后,在生成氢气的过程中,根据电极材料的固有属性以及其表面的电子特性,我们可以将阴极的析氢反应过程分为两种,一种是Heyrovsky反应过程,即吸附态的氢(H*)直接与电子和氢离子相结合生成氢气(酸性电解液)或者吸附态的氢(H*)直接和水发生反应生成氢气(碱性电解液),这种H2的电化学脱附机理我们称之为Volmer-Heyrovsky机理;另一种是Tafel反应过程,由两个吸附态的氢(H*)在电极上直接反应生成氢气,这样一种H2化学脱附机理我们称之为Volmer-Tafel机理。
整个阴极反应的详细过程可以由如下的方程式来描述:
酸性电解液中[1]:
Volmer反应:H eminus; → H*
Heyrovsky反应:H* H eminus; → H2
Tafel反应:2H* → H2
碱性电解液中[2]:
Volmer反应:H2O eminus; → H* OHminus;
Heyrovsky反应:H2O H* eminus; → H2 OHminus;
Tafel反应:2H* → H2
1.1.3 催化析氢(HER)的主要参数
在电催化过程中,结合实验与计算,在评价催化剂电催化析氢时的参数主要有一下几个:过电势,塔菲尔斜率、氢吸附吉布斯自由能(△GH*)、电化学阻抗和电化学活性面积。通过对这几个参数的测试可以更好地评价催化剂的HER催化性能更加准确地呈现HER反应进度,在这里我们详细介绍一下电催化过程中过电势,塔菲尔斜率、氢吸附吉布斯自由能(△GH*)几个参数。
在HER反应过程中,由于反应存在反应活化能垒,为了克服这些能垒,因此在电化学过程中,HER反应的电位很难等于热力学平衡时的平衡电位。所以在反应过程中,我们还需要额外多加一部分电压来克服反应的能垒。这一部分电压即为反应时的过电势。过电势的大小是评判HER催化性能好坏的重要标准之一。过电势越小,说明能量的利用率就会越高,催化性能就越好。随着电流密度的增加,过电势也会逐渐增加。在实验中,人们经常取电流密度为10 mA cm-2时的过电势进行比较。
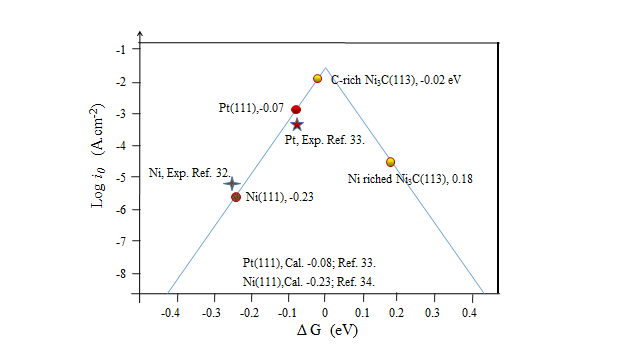
Tafel在1905年提出的经验公式:ƞ = a blog| j |,描述了电化学反应过电势与电流密度之间的关系,其中b是Tafel斜率,j是所测的电流密度。当过电势为零时,我们可以计算得到交换电流密度(j0),当b的数值越小时,| j0 |的值越大,表明电子转移速率更快,HER的性能越好。
在本文中,我们主要计算了氢原子在Ni3C(113)面的氢吸附能以及氢吸附吉布斯自由能(△GH*)。在催化过程中,氢在催化剂表面存在吸附与脱附两个过程,因此为了探究催化剂表现活性位点对于氢原子的吸脱附性能,我们需要对其氢吸附能进行计算。由于催化的过程是吸附与脱附两个过程,因此活性位点对于氢原子的吸附能要小于零,否则不会对氢原子产生吸附。同时,活性位点的氢吸附能的绝对值不能过大,因为过大的氢吸附能会使得氢原子的脱附过程变得十分困难。此时,氢气会覆盖在催化剂的表面阻止电极和氢离子的接触,使反应很难继续进行。所以,理想的催化剂活性位点必须有一个合适的氢吸附能,保证HER反应能够快速持续的进行。
通过氢吸附能,我们可以计算出催化剂的氢吸附吉布斯自由能(△GH*),当△GH*的绝对值接近于0时,此时log| j0 |的值最大。因此当| △GH* |越趋近于零,催化剂的催化性能越好。
除此之外,我们还能够利用“d能带中心值(d-band center)”这一描述符对HER性能进行一个简单的判断。对于过渡金属来说,sp轨道上的电子经常是半满的,因此能带展宽非常大,为了简化,我们可以认为所有sp带的贡献都是一样的,此时我们只需要考虑d带的贡献即可。
在过渡金属中,当d轨道与吸附物能级发生相互作用后,会使得能级发生分裂,当两个能量不同的原子轨道进行重叠成键,形成的成键轨道会比原来的能级轨道还要稳定,而同时形成的反键轨道会比原来不稳定的能级能量还要高,当反键轨道没有填充时,整个吸附体系能量会降低,可以稳定的吸附过渡态分子。当d轨道电子较少时,此时,费米面下降,即d能带中心上升,使得更多的反键轨道浮出费米面,进而增强了材料对于过渡态分子的吸附能力。但是并不是d轨道电子越少越好,因为过强的吸附能力可能会导致吸附分子之后的脱附过程难以完成,进而阻碍了催化作用[3]。据此Norskov提出了d能带中心值的概念,也可以理解为d电子的平均能量,值越正,可认为具有越强的催化能力。其计算公式如下
epsilon;d
d能带中心对于解释过渡金属催化剂的活性有很强的指导意义,但他的使用条件是必须具有d能带电子,因此对于Ni基化合物,使用d能带中心值是十分合适的。
1.2非贵金属析氢催化剂的研究进展
一般来说,基于铂、铱和钌等贵金属的反应催化剂具有令人满意的催化速率[4, 5],但是,这些贵金属催化剂稳定性差、成本高,严重制约了其大规模应用。因此,开发高性能、低成本的催化剂来替代这些贵金属已成为清洁能源研究的一项重要工作。为此,科研工作者们将自己的目光转移到了非金属催化剂,过渡金属化合物等极具催化潜力的材料上面。现如今,二维材料作为一种新兴的纳米催化材料,相比于其他纳米材料,在一定的电催化剂质量负载条件下暴露的电催化活性位点更多,而且其简单有序的分子结构使得从理论和实验角度识别活性中心变得更加容易,成为了贵金属基电催化剂的理想替代品。
1.2.1非金属催化剂
早在2009年,Dai等人就发现发现氮掺杂碳纳米管(N-CNT)有望在ORR应用中替代铂基催化剂进行催化反应[6, 7]。从那时起,用于各种电催化过程的2D非金属催化材料的开发已经取得了巨大的进展。这些材料包括:石墨烯基材料、类石墨烯碳氮化物(g-C3N4)、六方氮化硼(h-BN)和黑磷(BP)[8-11]。
自2004年Geim和Novoselov发现石墨烯以来,石墨烯已经成为电子、生物医学工程和电催化等各个领域的研究热点。石墨烯是一种单原子厚度的二维碳晶体,其中以sp2杂化的碳原子被填充在类似于蜂窝的六角形晶格中。作为最基本的碳结构,它被认为是其他尺寸碳材料的基本构件,包括零维的富勒烯、一维的碳纳米管和三维的大块石墨或其他碳结构。石墨烯边缘和缺陷处的电子态与基底平面不同,能够显著提高电催化活性。Jiao等人利用理论计算得出的关键反应中间产物的吸附能与测量的电化学反应速率相互印证,研究了一系列非金属杂原子掺杂石墨烯在HER催化中的性能,是探究石墨烯基电催化剂原子尺度设计的先驱工作之一[8]。
氮化碳是科学文献中首次报道的人造聚合物之一,类石墨烯氮化碳(g-C3N4)是具有范德华层状结构的二维晶体,被认为是所有碳氮化物材料中最稳定的同素异形体。与石墨烯类似,在g-C3N4的晶体结构中碳原子和氮原子通过sp2杂化进行结合,可以看做是用氮取代碳的六方碳骨架。在g-C3N4催化剂的合成中,可以通过多种方式对活性中心进行调节,其目的是改变目标中间体的吸附能,使其更有利于HER反应。通过将g-C3N4和氮掺杂的石墨烯复合,可以达到与金属催化剂相当的HER性能[11]。
黑磷烯(BP)自1914年首次合成以来,已有100多年的历史。黑磷烯是具有正交晶体结构的层状半导体,一个磷原子与另外三个磷原子以共价键结合,在单原子层中形成起皱的蜂窝结构。这三个键占据了磷的所有三个价电子,导致带隙为~2eV,通过控制黑磷烯的厚度,我们可以对其带隙进行调整。然而,由于它在电催化条件下的稳定性差,电导率低,因此在电催化中的应用报道很少。但是,在最近的研究中,Luo 等人合成了一种0D-2D Ni2P和黑磷烯的异质结构复合材料,由于黑磷烯高的比表面积以及Ni2P与黑磷烯界面之间可调的电荷载流子浓度,使得该杂化材料具有优异的电催化性能。与工业应用的Ni2P相比,这种新合成的材料在酸性介质中具有更好的HER性能[12]。
1.2.2 金属硫族化物催化剂
金属硫族化物是最大的一类电催化材料,由于具其有优异的结构和电子性质,它们早已在HER催化中有着十分广泛的应用。金属硫族化合物按其二维结构形式可分为层状过渡金属硫族化物(TMD)和非层状金属硫族化合物(NMC),其中二维过渡金属硫族化物电催化剂的开发是合成低成本高性能的HER电催化剂最有潜力的方向之一。
过渡金属硫族化物一般可以用化学式MX2来描述(M是过渡金属元素,X是硫族元素如S或Se)。块状过渡金属硫化物由是由范德华力堆叠的单层组成的,而过渡金属硫化物单层的一般晶体结构可用三明治结构进行描述,其中一层过渡金属位于两层硫族元素之间(即X-M-X)。二维过渡金属硫化物的独特之处在于它们的多晶结构。例如,2H(六边形)、1T(三角形)和3R(菱形)是MoS2的三种常见晶体结构,它们取决于Mo和S原子之间的不同配位模型和层间的堆积顺序[13]。由于存在不同的晶体类型和带隙,使得同种元素组成的过渡金属硫族化物之间的表面性质不同,进而导致催化活性产生差异。
在电催化应用中,通过DFT计算和实验已经证明,过渡金属硫族化物的活性位点一般在层边缘处的X位而不是基面内的X位[14, 15]。因此,为了进一步提高TMD的电催化性能,人们开发了各种方法来暴露更多的活性边缘位点(如横向尺寸控制、纳米结构、缺陷工程等)。此外,位于基平面的M或X位点都可以利用杂原子掺杂进行激活。实验和DFT计算表明,金属原子(Fe,Co,Ni)和非金属原子(B,N,O)掺杂均能有效地提高过渡金属硫族化物的HER活性[16, 17]。
1.2.3层状过渡金属碳化物和氮化物(MXene)
2011年,一种新的2D纳米材料MXene被发现。Ti3C2Tx是第一个被合成出来的MXene材料[18],迄今为止,通过实验合成或理论预测已经证实了存在70多种不同的MXene型材料。MXene的一般化学式是Mn 1AXn,其中M代表过渡金属元素,A主要是IIIA或IVA族元素,X是C或N,n=1,2或3。MXene材料具有金属导电性和亲水性,非常适合于电催化。
像Ti3C2Tx这样的MXenes催化活性较低,因此很难直接用作电催化剂。但是在之后进行合成的MXene材料中,Mo2C却显示了较为优异的电催化性能[19]。Lu等人将Mo2C包埋在氮掺杂多孔碳纳米片中[20],这种材料的起始点位接近0 V,在10 mA cmminus;2 的电流密度下,过电势也仅仅只有45 mV,大大低于大多数报道的HER电催化剂的实验数值,催化性能与贵金属催化剂Pt相当。由于MXene是一种新型二维材料,因此针对MXene材料的研究还并不太充分,因此在MXene材料的HER催化研究中我们还有很长一段路要走。
1.2.4 过渡金属氢氧化物
很多传统的过渡金属氢氧化物(TMH)由于活性低和导电性差,与其他的电催化剂相比,其性能并不突出。但是,近年来许多研究表明,通过减小过渡金属氢氧化物的厚度,可以提高它们的电催化活性。因此,二维过渡金属氢氧化物成为了在HER催化应用中极具发展潜力的候选电催化剂之一。
层状过渡金属氢氧化物(LMHs),包括层状单金属氢氧化物(LSHs)和层状双金属氢氧化物(LDHs),是一类以无插层阴离子的金属羟基为主体的层状材料。目前,人们已经合成了各种二维LMH材料,其中大多数都表现出高效的电催化性能。在HER催化中,超薄Ni(OH)2与铂衬底结合的电极表现出协同效应,极大的促进了碱性HER过程[21]。在该杂化体系中,Ni(OH)2促进了水的吸附和解离,而Pt表面则促进了氢的重组与解吸。
1.3 实验材料介绍
通过上一节对于过渡金属基催化剂的介绍,我们可以知道过渡金属基催化剂是一种值得深入研究的HER催化剂。在这些过渡金属中,过渡金属镍储量巨大,价格便宜,由于镍基化合物在HER催化中有着十分高效的催化效果,因此被认为是一种极具发展前景的过渡金属催化剂。镍基过渡金属催化剂主要包含了磷化镍(NixPy)以及碳化镍(NixCy)两种化合物,由于这两种材料含量丰富、催化活性高、稳定性好、易于合成,因此在电化学催化领域引起了广泛的关注。
在过渡金属碳化物中,金属密堆积的主晶格中存在小的间隙碳原子,使得过渡金属碳化物具有间隙合金的性质,在催化和电化学领域引起了广泛的关注。在这些过渡金属碳化物中,Ni3C有着极为优异的催化活性和化学稳定性,在近年来也被广泛用于HER电催化的研究。在实验上,Wang等人利用无模板的自发泡策略制备出了一种将Ni3C纳米粒子嵌入多孔碳网络中的复合材料(Ni3C@PCN)[22]。实验测得该材料的起始电位为-65 mV,在50 mA cm-2的电流密度下的过电势为262 mV,酸性溶液中的Tafel斜率为63.4 mV/dec。进一步的对Ni3C材料进行改性,Fan等人合成出了氮掺杂碳纳米片与铁掺杂Ni3C纳米点的复合材料[23]。在碱性电解液中,该材料在10 mA cm-2的电流密度下的过电势为292 mV,Tafel斜率为34.6 mV/dec,也展现出了较好的电催化性能。从实验方面来看,Ni3C是一种非常有效的HER电催化剂。在计算方面,针对Ni3C的电催化研究并不多见,而且主要集中在了磷化镍方面,因此对Ni3C的电催化理论研究也是十分有必要的。
1.4 研究意义
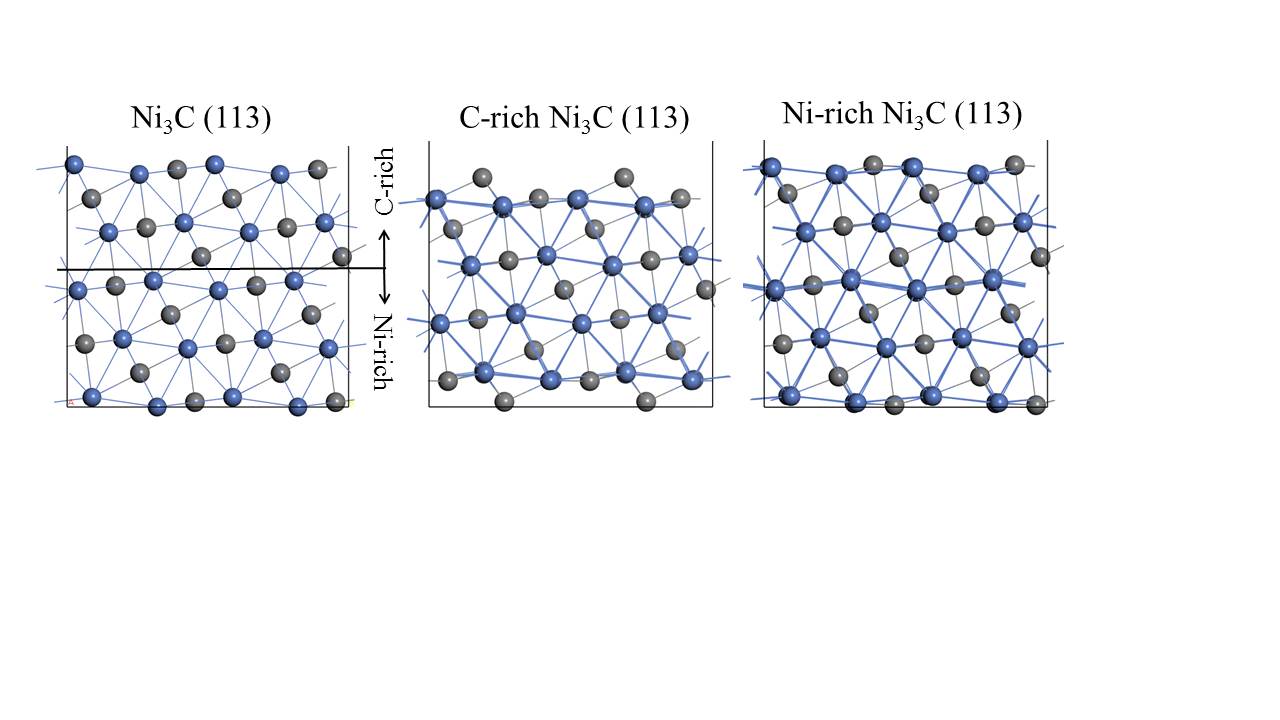
通过上一小节的介绍我们了解到,Ni3C是一种极为优秀的电催化材料,但是这些文献并未对Ni3C为何具有优异的HER电催化性能进行深入的解释。为了深入探究Ni3C电催化活性的根源,在本文中,我们选取了Ni3C(113)晶面作为研究对象,从理论方面揭示了Ni3C的催化活性来源。Ni3C(113)晶面上存在两种不同的截面,根据两种截面上碳与镍的含量比的不同,我们将其分为富含碳原子的Ni3C(113)晶面(C-rich Ni3C(113)晶面)以及含镍原子的Ni3C(113)晶面(Ni-rich Ni3C(113)晶面)。我们通过计算这两种截面的d能带中心值、氢吸附能以及氢吸附吉布斯自由能,对其HER催化性能进行评判。
第二章 实验计算原理
2.1 第一性原理
第一性原理最初仅仅只是一个哲学用语,主要用来代指任何基础原始能够自证的,仅包含假设与猜想,不可从其他已有的定理或是经验定理推导得到的理论。与经验定理不同的是,第一性原理体系有着自洽性与完备性,衡量一个理论的价值高低仅仅在于其描述是否符合事实。在物理学上,参考原子核与电子之间的相互作用和他们基本的运动规律,以量子力学原理为基础。根据实验中的具体要求将薛定谔方程近似处理之后求解得要我们需要的结果的方式叫做第一性原理。
正如第一段我们所介绍的,由于薛定谔方程在求解某些复杂的体系的时步骤过于繁琐,因此,在实际的应用时,我们需要对薛定谔方程进行近似处理。根据不同的近似条件,我们利用得到不同的近似理论,例如波恩-奥本海默(Born-Oppenheimer)绝热近似[24]、Hartree-Fock近似[25]、密度泛函理论(DFT)。在材料学中的第一性原理主要包含两大分支:密度泛函理论和分子动力学。在本文中,我们主要运用的是密度泛函理论进行,因此在接下来的小结我们会对密度泛函理论进行简要的介绍
以上是毕业论文大纲或资料介绍,该课题完整毕业论文、开题报告、任务书、程序设计、图纸设计等资料请添加微信获取,微信号:bysjorg。
相关图片展示: