基于深度测序的微生物组进化分析及物种溯源算法研究毕业论文
2020-04-04 10:59:06
摘 要
随着下一代测序技术的发展和基因数据库的完善,针对微生物的研究逐渐从传统的基于培养的方法转变为基于基因组学的宏基因组学方法。其中标记基因分析是主要的一类研究方法,OTU划分是这类方法的关键步骤。传统的OTU划分方法都采用直接计算序列间差异来表示序列距离,这种距离计算存在不可预见的错误,同时大部分方法也需要设定相似度阈值,而阈值的选择往往缺乏依据。我采用替换模型计算序列间的进化距离来作为序列距离,使用无需相似度阈值作为输入的泊松树过程模型来划分OTU。在对两个已知完整真实序列的模拟群落的测试中,相比UCLUST和UPARSE方法,基于泊松树过程模型的无监督OTU划分方法在OTU多样性上获得了与真实物种更接近的结果,并且在OTU序列质量上能更加还原真实序列。
关键词:OTU划分;泊松树过程模型;宏基因组学;系统进化树;标记基因分析
Abstract
With the development of next-generation sequencing technologies and the improvement of gene databases, the research on microorganisms has gradually shifted from a traditional culture-based approach to a genomics-based metagenomics approach. Among them, marker genes analysis is the main approach of metagenomics. OTU-picking is the key step of this method. The traditional OTU-picking method directly calculate difference between sequences as the distance between pair sequences. This distance has unforeseen errors. I calculate the evolutionary distance between the sequences using nucleotide substitution model as the sequence distance, and construct OTU using a Poisson tree process model that does not require a similarity threshold as input. This unsupervised OTU-picking method based on the Poisson tree process model obtained more accutate results in OTU diversity and better OTU sequences with higher quality than the UCLUST and UPARSE methods on two mock microbial communities dataset.
Key Words:OTU-picking;Poisson tree process model;metagenomics;phylogenetic tree;marker genes analysis
目 录
第1章 绪论 1
1.1研究背景 1
1.2 国内外研究现状 2
1.2.1 OTU介绍 2
1.2.2 划分OTU的原因 3
1.3.3 常见OTU划分方法 3
1.3.4 OTU划分的现存问题 5
1.3 研究目的及意义 6
第2章 基于PTP模型的OTU划分方法 7
2.1 系统进化树 7
2.2 PTP模型 8
2.3 划分方法 9
2.3.1 方法思路 9
2.3.2 方法流程 10
第3章 方法测试 12
3.1 测试数据集 12
3.2 实验设计 13
3.3 测试结果及分析 14
3.3.1 OTU多样性评估 14
3.3.2 OTU序列质量评估 15
3.4 测试总结 17
第4章 结论 19
4.1总结 19
5.2展望 19
第1章 绪论
本章主要从研究背景、研究目的、研究意义、国内外研究现状、毕业设计研究内容及预期目标几个方面进行阐述OTU、常见OTU划分方法和现存的问题。
1.1研究背景
微生物无处不在,包括细菌,古菌以及一部分的真核生物,它们对地球的生态系统是至关重要的,几乎所有宏观动植物都有与之寄生共存的微生物。生命过程中,转化生命的关键必须元素碳、氮、氧和硫到生物体的化学循环主要依靠微生物完成,所有的动植物都要紧密依赖于特定的微生物群落来获取必要的营养、金属元素和维生素。在人体肠道和口腔中的微生物群落能促进我们获取食物中的能量,如果没有它们,我们就无法充分消化食物,它们还保护我们免受一些致病因子的侵害。研究微生物有利于改善生态环境和维持人体健康。
对于宏观动植物的研究,我们能够培养不同物种的个体,轻易的获取个体全基因组信息。但环境中的微生物往往是以群落的形式存活,微生物对环境的影响和微生物的功能也是在复杂的群落内进行的。这些群落是复杂而平衡的整体,能迅速灵活的适应环境变化。在实验环境下,培养总是有利于最能在实验条件下繁殖的微生物(俗称“实验室杂草”),而不一定是环境中数量占优的微生物。在1985年,Staley和Konopka就开始研究有多少种微生物能够从自然环境移到实验环境单独培养,随后的研究表明绝大多数通过各种染色方式能在显微镜下观察到存活状态的微生物都无法单独诱导到培养皿上长成菌落或者在试管中培养,研究估计在土壤环境中只有0.1%-1.0%的细菌能在标准环境下单独培养,而水生环境中的可培养细菌可能会相对于土壤少10-1000倍[1]。这虽然是有争议的数值,但微生物难以单纯培养研究是普遍认可的事实。
微生物物种繁多,存在众多未知物种,对每一种物种进行单独培养、基因测序几乎是不可能的。因此,传统的基于培养的研究无法用于微生物中,针对微生物研究的宏基因组学便获得了快速的发展。宏基因组学直接从微生物环境样品中提取全部DNA,构建宏基因组文库,再利用基因组学的技术对环境样品中的所有微生物的遗传组成和环境的微生物群落功能进行研究。一般环境中的微生物的遗传组成和群落功能仅仅是能被抽样出来,但无法进行准确识别和描述,宏基因组学中的“宏”就要求我们开发出计算方法来尽可能的理解这些遗传组成和群落功能。此外“宏”还意味着我们要在整体水平上研究微生物,超越个体生物水平,关注群落中的基因,研究基因是如何影响群落的功能的。
这种不依赖于培养的识别环境中的微生物的组学方法变得越来越重要。宏基因组学分析环境微生物群落的组成和功能时,通常会先对某个标记基因进行分析。目前被最广泛使用标记基因的是核糖体RNA(rRNA),而16s rRNA是唯一一种广泛用于微生物识别、分类的标记基因。核糖体RNA是核糖体的关键组成结构和功能成分,它是细胞内蛋白质合成的场所,能读取信使RNA核苷酸序列所包含的遗传信息,并使之转化为蛋白质中的氨基酸以合成蛋白质。所有的生物都有rRNA,并且rRNA有足够信息用来区分不同的物种和测量物种之间的进化距离。Pace最早使用核糖体RNA来研究环境微生物群落[2]。核糖体RNA相对较为保守,不然容易发生突变。核糖体RNA是研究微生物群落组成和微生物群落之间关系最重要的工具,用于了解宿主或环境的状态。最常用的标记基因是16s rRNA,其他常见标记基因有18s rRNA、ITS等。
标记基因分析至关重要的第一步通常是给目标测序数据集划分出基本的多样性单元。理想情况下,我们希望划分的多样性单元能够与样本中的真实微生物的物种分类单元对应。微生物测序通常是对整个环境样本中的某个标记基因进行测序,测序结果包含了所有的序列,为了后续研究的方便,我们需要从这些序列中选取特定的多样性单元用于后续样本多样性、微生物群落组成和功能的分析。如何定义一个具体的最具生物学意义的多样性单元还存在争议,目前最常用的定义是可操作分类单元(operational taxonomic units,缩写为OTU)。OTU定义为距离相近或相似度高的标记基因序列的集合。下一节描述OTU的详细定义、如何定义序列的距离或相似度、常见的OTU划分方法和存在的问题等。
1.2 国内外研究现状
1.2.1 OTU介绍
标记基因分析对微生物测序序列分析的第一步就是OTU划分。我们通常希望在物种水平对测序序列进行OTU划分。Sneath和Sokal最早引入了可操作分类单元(OTU)的概念[3]。OTU通常定义为:使用聚类方法根据特定的序列相似度阈值划分得到的生物序列的集合。划分OTU的目的是根据可观测特征将生物序列定量划分相同组中,这里的分析中,特征就是基因序列,我们希望尽可能根据他们之间的进化关系来对生物进行分层分类。用不同的阈值划分出来的OTU可以近似的代表不同水平的物种分类单元(种、属、科等),在16s rRNA序列研究中,广泛采用97%作为种水平OTU划分的相似度阈值[4]。但最近有研究表明对16s rRNA全长序列进行物种水平的OTU划分时,相似度阈值应采用98.5%[5]。
1.2.2 划分OTU的原因
划分OTU有下面几个原因。第一,OTU定义了类似物种的一个可操作的集合,方便后续的分析。第二,OTU能将原本环境中属于同一菌株但发生变异的序列划分到同一个集合中。细菌染色体经常包含多个16s基因的拷贝,旁系同源(生物体中的某个基因被复制了,那么两个副本序列就是旁系同源)之间的16s rRNA序列差异非常小,聚类会将这些旁系同源分到同一个OTU。这样使得划分结果在大多数情况下是合理,一个性状对应一个OTU。第三,OTU划分也能将由同一微生物物种形成的不同菌株的序列分到同一个集合中。一个微生物物种可能形成多个不同的菌株,这些菌株也由于旁系同源会有不同的16s rRNA序列,这样的旁系同源导致了物种内的多样性。与上一种情况不同的是,通过调整参数,OTU聚类也能将这样的多个菌株合并创建一个或者是相对少的OTU来表示一个物种。但如果目标是给每个性状划分一个OTU,这样的划分可能不正确,因为不同菌株之间在性状上有显著差别。此外,如何定义一个微生物物种还有一定争议,有些研究支持用菌株来定义微生物物种。因此不一定能够按一个物种一个OTU的目标来划分OTU。第四,OTU划分去掉大部分由实验产生的错误序列。扩增测序各个过程特别是PCR扩增很容易产生错误序列,这些序列都是没有生物学意义的。常用的OTU聚类方法都会通过一些流程筛掉这些错误序列。
1.3.3 常见OTU划分方法
目前OTU划分方法主要有三类,依赖参考数据的聚类方法(closed-reference clustering)、从头聚类方法(de novo clustering)和两种方法的混合(open-reference clustering)。OTU划分的三类方法如图1.2所示,图片来自Navas-Molina[6]。
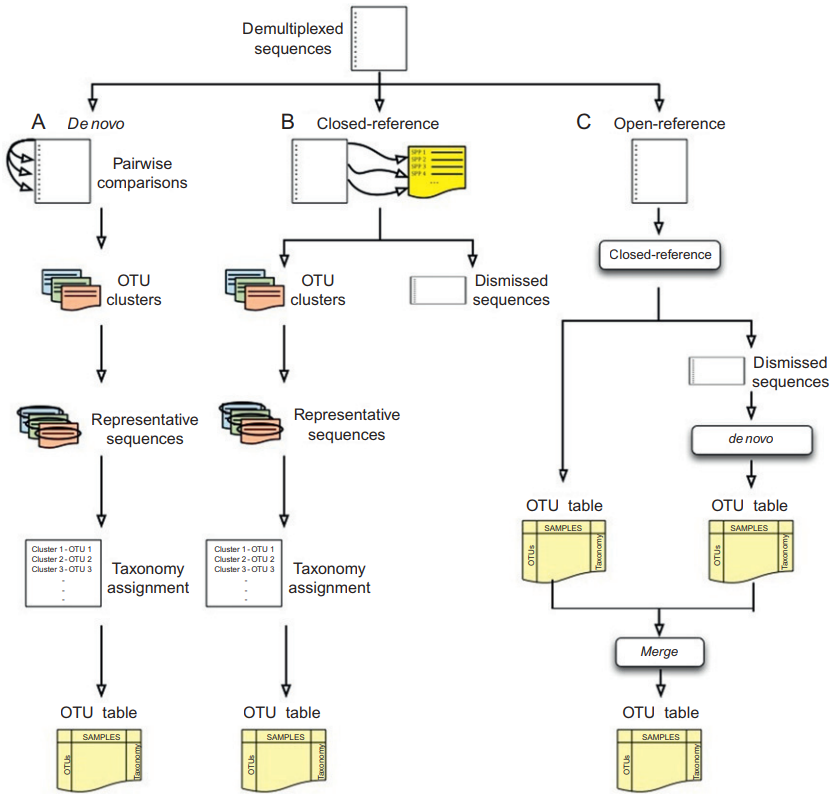 图1.2 OTU划分的三类方法
图1.2 OTU划分的三类方法
closed-reference clustering方法将测序序列与参考数据库中的序列进行比较。计算测序序列与参考数据库中序列的距离,并以参考序列为OTU代表序列,将测序序列分配到与之距离最近的参考序列中去。这种划分OTU的方法的效果取决于参考数据库的序列是否能够充分代表真实环境中的序列,如果测序序列中有大量未知的序列,那么这些无法匹配到参考数据库的未知序列就被不会分配到任何OTU,而是被丢弃。此外,通常只选相互相似度小于97%的参考序列作为OTU代表序列,但这个97%阈值还存在争议。这种划分方法存在明显的问题。首先,通常认为相似度高于97%的两个测序序列来自同一个物种,但这种方法可能会出现这样的情况,分配到同一参考序列的两个测序序列之间的相似度小于97%。其次,一个测序序列可能与多个参考序列的相似度高于97%且相等。closed-reference clustering方法的优点有聚类速度快,提高参考数据库的质量就能显著提升OTU划分的质量。
de novo clustering方法不依赖于任何参考数据,从头对测序序列进行聚类。这类方法计算测序序列之间的距离用于聚类。主要有级联聚类(ESPRIT-Tree[7])、基于距离的贪婪聚类(UCLUST[8])和基于丰度的贪婪聚类。与closed-reference方法的速度相比,级联聚类的计算时间通常是和序列的数目成二次关系,但ESPRIT-Tree方法做了速度优化,时间复杂度降为拟线性时间。测序通量扩大和测序错误会增大唯一序列的数量,导致需要大量的内存和时间来进行聚类。但通过质量控制等预处理可以有效减少唯一序列的数目,克服这个问题。而贪婪聚类方法通常是通过以指定相似度阈值来构造一系列中心序列的方式对序列进行聚类。以UCLUST为例,UCLUST算法以固定顺序输入序列,每输入一个序列就判断该序列是否与已存在的某一中心序列相似度高于固定阈值,若高于固定阈值则分配到那个中心序列,否则该序列就成为一个新的中心序列。如果与多个中心序列的相似度阈值都高于固定阈值,就分配给相似度最高的第一个中心序列。最后在把分配到同一个中心序列的序列当成一个OTU集合,每个中心序列就是对应的OTU代表序列。显然,这样的贪婪聚类方法会受到输入序列的顺序的影响。
open-reference clustering方法就是将前面两种方法混合使用。先使用closed-reference clustering方法对序列进行聚类。然后对无法与参考数据库匹配的剩下序列用de novo clustering方法聚类,最后将两种方法生成OTU合并。目前一些标记基因分析集成平台就是采用这样的方法,例如QIIME[9]和Mothur[10]。
1.3.4 OTU划分的现存问题
目前普遍认为de novo clustering方法要优于closed-reference clustering和open-reference clustering,而OTU划分的好坏直接对后续分析的结果造成显著的影响。但是目前这些方法都存在一些问题,主要有序列距离或相似度的定义问题和阈值选择缺乏依据的问题。首先对于序列相似度的定义,目前大部分方法都采用直接计算序列之间的差异来表示序列之间的距离。通常先进行序列比对,然后对比对后序列计算相似度。用不同的方法来计算距离会得出不同的结果。CD-HIT聚类定义相似度为:相同位点数目与短序列长度的比值。另一类是BLAST中的定义:相同位点数目与比对长度的比值。这种序列的直接差异忽略了在进化过程中,不同物种之间序列核苷酸的替换次数、核苷酸出现的频率以及核苷酸发生替换的概率,会错估真实的物种序列距离。对于阈值的选择问题,大部分de novo clustering方法和closed-reference clustering方法都需要一个相似度阈值来进行聚类。通常物种水平的聚类选用97%作为相似度阈值。但有研究表明,阈值的微小改变就会使有些聚类方法的OTU划分结果有显著变化[11]。虽然已经有一些方法(CROP[12])不采用固定阈值,而是软阈值,即聚类过程中阈值会改变。但不同基因的保守性是不同的,16s rRNA基因的不同区域的保守性就完全不同。有研究表明,如果使用全长的16s rRNA,应采用98.5%作为物种水平的相似度阈值[5]。因此,使用固定阈值或软阈值的聚类方法的阈值选择都缺少依据。
1.3 研究目的及意义
本研究的目的是克服OTU划分方法的现存问题,提出一种无需指定相似度阈值和有更好的序列距离定义的OTU划分的新方法。为了更好的定义序列之间的距离,我从系统进化分析中获得启发,采用序列的进化距离来表示序列之间的距离。然后用进化距离来构建系统进化树,再使用泊松树过程模型对系统进化树上的序列进行划分集合,每一个集合表示为一个OTU,集合中丰度最高的序列作为OTU代表序列。我基于这种方法开发一套不依赖于参考序列的基于泊松树过程模型的无监督OTU划分流程。这种新方法优化了序列距离的定义,使距离计算更贴近真实情况。同时由于采用最大似然估计的统计方法进行系统进化树的构建和序列集合的划分,而不是聚类,这种方法也无需选择阈值,从而克服了聚类方法的阈值选择缺乏依据的问题。用这种方法获得的OTU具有更好的生物学意义,有利于后续样本多样性、微生物群落组成和功能的分析。
第2章 基于PTP模型的OTU划分方法
本章描述基于泊松树过程模型(Poisson tree processes model,缩写为PTP)的OTU划分方法的思路和划分流程。描述这种方法前,我将对系统进化树和PTP模型进行介绍。
2.1 系统进化树
基于泊松树过程模型的OTU划分方法虽然不需要输入相似度阈值参数,但需要先对序列构建系统进化树。系统进化树是表示物种或基因间系统发育关系的树状图,分支长度表示进化距离的差异,叶子节点表示物种或基因,内部节点表示分支事件。随着测序技术的发展,系统进化树几乎应用到了生物学的各个分支中。系统进化树是一种能解决很多生物学问题的重要工具。图2.1展示一个包含三个域的系统进化树,它以不同颜色的子树表示自然界中的三个域:细菌域、古菌域和真核生物域。由此可以进行拓展,如果对大量序列构建系统进化树,然后在物种水平进行划分,我们就可以得到序列在物种水平的集合。采用泊松树过程模型划分OTU正是基于这样的思想。
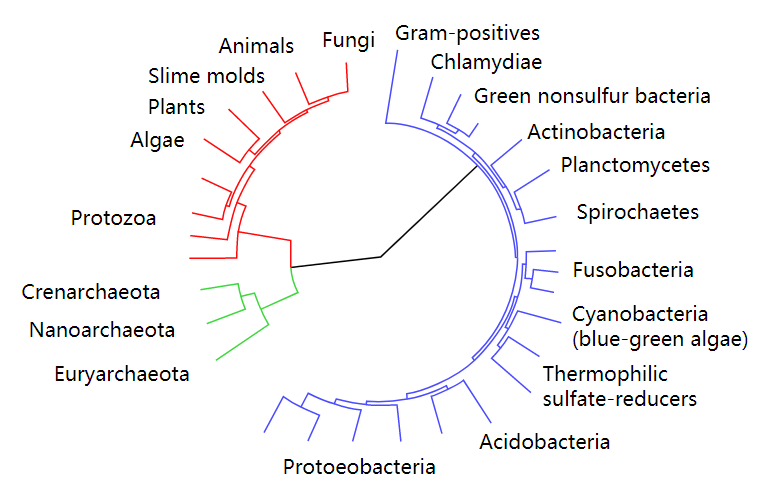
图2.1 按域进行划分的系统进化树
系统进化分析通常采用替换模型来表示物种序列之间的进化距离,然后再用聚类或统计的方法来构造系统进化树。目前系统进化树的构建方法分为两类,一类是基于距离的方法,另一类是基于特征的方法。基于距离的方法通常是聚类法,聚类法速度快,能处理大规模数据,但无法处理距离远的物种。基于特征的方法通常是统计的方法,主要有最大简约法、最大似然估计法和贝叶斯方法,这类方法能使用更复杂的替换模型来计算序列之间的进化距离,因此的得到的结果比基于距离的方法更好,但是计算复杂度较高,其中贝叶斯方法速度最慢。构建好的系统进化树非常困难,目前还没有有效的公认方法来评价哪种构建方法最好。在计算时间和结果优劣的权衡下,我选择最大似然估计法来构建系统进化树。
2.2 PTP模型
泊松树过程模型(PTP)是2013年提出的,用于在系统进化分析中划分物种的模型[13]。PTP模型只需要输入一颗完整的系统进化树,就可以对叶节点的序列进行划分。PTP模型直接用核苷酸替换次数来表示物种产生的速率,而核苷酸替换次数可以由系统进化树的分支长度求得。两个节点之间的分支长度表示序列每个位点的平均替换次数,用替换次数的大小来区分种间和种内差异。为了使这种区分合理,必须先做基础假设:物种间的替换次数显著多于物种内的替换次数。
假设每次替换有小概率产生新物种,且每次替换是独立的,替换是发生在离散的一小段时间内,时间间隔非常小,因而只发生一次替换。在这样假设下,在κ次替换之后,产生η个物种的概率服从二项分布。因为我们假设每次替换有小概率ρ产生新物种,替换发生的次数比物种数η大得多,所以这个过程在连续时间内是按频率来服从泊松分布,因此,直到下一个物种产生之前,替换数将服从指数分布。PTP方法可以方便的从系统进化树中的分支长度获得替换数。



