FASTQ格式文件压缩研究毕业论文
2021-03-15 20:14:03
摘 要
对下一代测序技术产生的海量DNA测序数据进行经济性存储和传输的最佳方式是对DNA测序数据的标准存储方式FASTQ格式文件进行压缩处理。为了能够对这方面的研究有所帮助,本文进行了以下工作:
- 对目前主流FASTQ压缩算法进行了压缩性能、运行时间方面的对比分析,选取压缩率最高、运行时间最长的LFQC算法作为要深入研究的对象;
- 选取MPEG高通量测序压缩工作组编制的标准梯度数据集,对LFQC算法的运行时间和压缩率进行详细测试;
- 分析测试结果,给出LFQC的优化方并解决算法中出现的部分问题。
实验结果表明LFQC无法对过大的FASTQ文件(碱基大小超过2Gb)进行压缩,可以将调用的通用压缩工具lpaq8替换为zpaq来解决这个问题;替换zpaq版本为715可以减少压缩消耗的时间。
本文的特色在于分析研究了目前压缩率最高的FASTQ无损压缩算法LFQC,并对其进行了优化,修复了诸如无法压缩大文件的问题,有助于研究人员对LFQC算法的使用,对下一代测序数据的存储和传输有重大意义。
关键词:DNA测序;下一代测序;无损压缩;FASTQ文件;LFQC算法
Abstract
The best way to economically store and transmit massive amounts of DNA sequencing data from next-generation sequencing technology is to compress the standard storage method FASTQ format file for DNA sequencing data. In order to be able to help in this area, this paper has done the following work:
- The mainstream FASTQ compression algorithm is compared with the compression performance and running time. The LFQC algorithm with the highest compression rate and the longest running time is selected as the object to be studied deeply.
- The standard gradient data set of MPEG high throughput sequencing compression group is selected, the run time and compression rate of LFQC algorithm are tested in detail.
- Analyzing the test results, giving the LFQC optimization side and solving some of the problems in the algorithm.
The experimental results show that the LFQC can not compress the oversized FASTQ file (the base size exceeds 2 Gb). You can replace the calling generic compression tool lpaq8 with zpaq to solve this problem. Replacing the zpaq version 715 can reduce the compression time.
The characteristics of this paper are to analyze and study the LFQC with the highest compression rate of FASTQ, and optimize it, such as unable to compress large files, help the researchers to use LFQC algorithm, the next generation of sequencing Data storage and transmission of great significance.
Key Words:DNA sequencing; next generation sequencing; lossless compression; FASTQ file; LFQC algorithm
目 录
第1章 绪论 1
1.1研究背景 1
1.2 国内外研究现状 2
1.3 研究目的及意义 4
1.4 课题研究内容 4
第2章 常用FASTQ压缩算法介绍 5
2.1 FASTQ格式 5
2.2 压缩算法 5
2.3 研究算法选取 9
第3章 LFQC算法分析 11
3.1 源码实现思路 11
3.2 算法拆分详解 11
3.2.1 原文件分割重组 11
3.2.2 分割文件处理 11
3.2.3 文件打包 14
3.3 压缩工具使用 14
3.3.1 通用压缩工具 14
3.3.2 工具选择分析 16
3.4 算法现存问题 16
3.4.1 输入文件名限制 16
3.4.2 本地运行错误 18
第4章 LFQC分步测试 20
4.1 数据集准备 20
4.1.1 数据集选取 20
4.1.2 数据集下载 20
4.1.3 数据集校验 21
4.2 测试过程 21
4.2.1 原始算法 21
4.2.2 替换lpaq8为zpaq 22
4.2.3 修改zpaq调用参数 22
4.2.4 替换zpaq版本 24
4.2.5 二次压缩 24
4.3 结果及分析 25
第5章 结论 29
5.1总结 29
5.2展望 30
绪论
本章主要从研究背景、研究目的、研究意义、国内外研究现状、课题研究内容及预期目标几个方面进行相关的阐述。
1.1研究背景
从1977年Sanger[1] 发明基于荧光标记的末端终止法DNA测序技术(第一代DNA测序技术)以来,DNA测序技术被广泛应用于生物和医学领域。2005年Margulies等人[2] 提出基于循环阵列合成的高通量测序(High-throughput sequencing,HTS)方法,又称为“下一代测序(next generation sequencing,NGS)”方法。该方法在各种实现平台上获得了超大量的DNA序列数据,在降低测序成本的同时,还大幅提高了测序速度,并且保持了高准确性。下一代测序以其无与伦比的通量、扩展性和速度,让研究人员以前所未有的水平研究生物系统,其发展极大地促进了基因组分析、遗传病诊断、食品安全、药物研发、育种等领域的研究。当今复杂的基因组学研究问题需要的信息深度已超过传统DNA测序技术的能力。下一代测序填补了这一空白,并成为解决这些问题的日常研究工具。
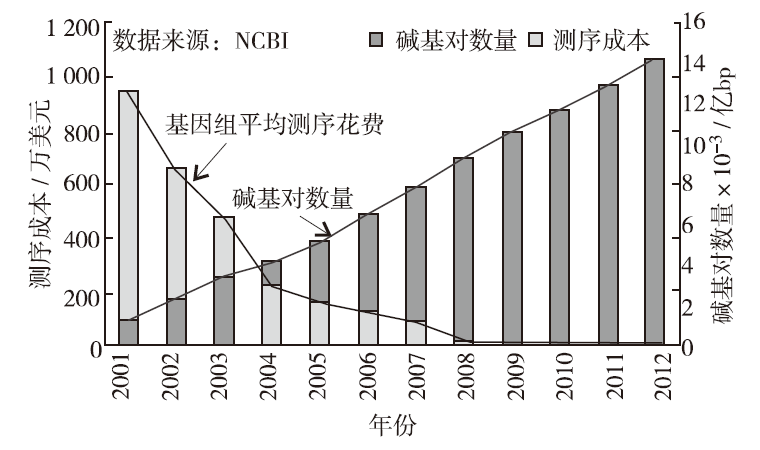 测序成本的降低和测序效率的提高,促使人们不断加强对DNA测序的大规模研究,使得DNA测序数据呈爆炸性增长(图1.1)[3] 。全球最大的DNA测序中心NCBI和EBI
测序成本的降低和测序效率的提高,促使人们不断加强对DNA测序的大规模研究,使得DNA测序数据呈爆炸性增长(图1.1)[3] 。全球最大的DNA测序中心NCBI和EBI
图1.1 DNA测序成本和序列数据量变化趋势
已经分别存储了超过万亿个碱基对,许多大型DNA研究项目,如国际癌症基因组计划、千人基因组计划、DNA元件百科全书计划和孟德尔遗传疾病计划等,正在以前所未有的速度产生海量数据。计算机是用于存储和处理DNA数据的主要工具,其微处理器性能和存储设备容量平均18~24个月翻一番,而DNA测序数据平均4~5个月就翻一番,DNA测序数据的增长速度已经远远超过计算机微处理器性能和存储设备容量的增长速度。
高通量测序数据生成的当前趋势表明,存储、传输和带宽成本将很快超过测序成本,成为基因组学和高通量测序数据应用于精准医学的主要瓶颈。下一代测序技术在改革基因组研究的同时,也带来了巨大的挑战。如何存储和传输高通量DNA 测序产生的数据,成为决定DNA 研究发展的重要因素之一。下一代测序数据容量的增长速度远远超过了存储成本降低和网络带宽增加的速度,由于其庞大的数据大小,存储和传输原始数据显然是不可行的。因此研究人员致力于压缩工具的开发,对原始数据进行压缩处理,显著降低高通量测序数据的大小以解决存储困难和传输缓慢的问题。
高通量测序数据通常以FASTQ格式[4] 或SAM(Sequence Alignment Map)格式进行存储。FASTQ是一种用于存储生物序列(通常为核苷酸序列)及其测序质量得分信息的文本格式。 为了简洁,序列字母和质量得分均用单个ASCII字符编码。该格式最初是由维尔康姆基金会桑格研究所(Wellcome Trust Sanger Institute)开发,旨在将FASTA格式序列及其质量数据集成在一起。SAM格式是一种基于文本的格式,用于存储与Heng Li[5] 开发的参考序列比对的生物序列。它广泛用于存储由下一代测序技术生成的数据,如核苷酸序列。FASTQ格式和SAM格式都具有较大的内存占用,虽然可以在文献中可以找到的大量通用压缩算法,但这些已被证实对测序数据进行压缩并不能达到一个很好的效果。因此,降低高通量测序数据存储、带宽和传输负担的一种方法是使用专为高通量测序数据开发的高性能压缩方法。
目前已经开发出的专用于高通量测序数据的压缩方法有很多,如DSRC2[6] 、FQC[7] 、Fqzcomp、Fastqz[8] 、Slimfastq、LFQC[9] 等。与通用压缩方法将数据视为简单的纯文本文件不同,这些专业的高通量测序数据压缩方法首先执行一种形式转换(读数标识符标记化或2位核苷酸编码),然后进行统计建模和熵编码[10] 。这些主流的压缩方法性能各不相同,它们的性能分析比较,最优方法具体的实现方式、最佳的应用场景及可以优化的地方都是值得我们探讨的地方。本文主要对这些问题进行深入研究。
1.2 国内外研究现状
由于DNA数据与一般的文本、图像、视频等格式不同,它只包含四种碱基符号{A,G,C,T},若将其看成随机字符串,则每个碱基符号需要2bits(),故对DNA数据进行压缩,每个碱基需少于2bits存储才能达到有效的压缩结果。传统的压缩算法如gzip[11] 、bzip2、7-Zip等虽然可以用来压缩测序数据,但是其每个碱基的存储空间大于2bits[12]。因此,研究人员致力于专用于DNA数据的压缩算法的研究。
2010年Tembe等人提出的基因组序列和质量数据的紧凑编码G-SQZ(Genomic SQueeZ)[13] ,是一种基于霍夫曼编码(Huffman, 1952)的排序读取特定表示方案,可以在不改变相对顺序的情况下压缩数据。G-SQZ在基准数据集上实现了65%至81%的压缩,并且无需从头扫描和解码即可进行选择性访问,可以被进行多线程并行计算应用程序所利用。它在组合碱基和各自的质量上使用零级霍夫曼编码,无法处理具有可变长度读数的数据集。
2011年Deorowicz等人提出的算法,其C 实现为DSRC(DNA Sequence Reads Compressor)[14] ,兼容分别由Sanger[4] 和加利福尼亚大学综合基因组生物学研究所(Institute for Integrative Genome Biology of University of California)提供的两种标准化格式的FASTQ格式。该算法处理DNA读数而不是基因组序列,它将FASTQ格式识别为有序的记录集合,并对记录进行数据流(分别为标题、DNA序列和质量评分)独立处理。标题数据流使用增量编码(Incremental Encoding)或统计编码处理后应用零阶霍夫曼模型,DNA序列数据流使用基于LZ77的压缩算法,质量评分数据流在使用统计编码的基础上进行霍夫曼编码。其压缩效率和性能明显优于处理相同数据格式的G-SQZ算法。
2012年,Jones等人提出了Quip 算法[15] ,基于统计模型,使用算术编码,可以对FASTQ和SAM/BAM 格式的下一代测序数据进行无损压缩。算术编码是对霍夫曼编码的改进,主要的优点在于可以使用非整数位对每个字符进行编码。如果某个字符出现的概率是0.1,可以使用理想的编码长度3.3。对于FASTQ 的不同行,使用不同的统计模型,这样就能取得更高的压缩比。尽管有这些优点,但是实际应用中却由于算术编码的专利权的限制,没有霍夫曼算法使用广泛[16]。



