FeN4位附近局部碳结构在提高氧还原催化活性中的作用外文翻译资料
2022-08-08 11:55:33

英语原文共 6 页,剩余内容已隐藏,支付完成后下载完整资料
FeN4位附近局部碳结构在提高氧还原催化活性中的作用
摘要:开发有效的非贵金属和氮共掺杂碳催化剂用于氧还原反应(ORR)需要对其催化活性的机制有一个基本的了解。 在本研究中,我们采用第一性原理密度泛函理论计算来预测ORR在三个FeN4上的一些关键参数(如Ominus;O键断裂的活化能和作为电极电位函数的自由能演化4具有不同局部碳结构的类型活性位点。我们发现,由八个碳原子包围的FeN4位在微孔边缘具有最低的O键断裂活化能(约0.20 eV),在促进直接四电子ORR的三个FeN4型活性位之间。 因此,我们的计算结果表明,在非贵金属催化剂中引入微孔可以通过促进FeN4minus;C8的形成来提高其对ORR的催化活性,其具有高比活性的活性位点。
- 导言
一类热解过渡金属和氮衍生的碳材料(表示为TM-N-C)已被证明是在酸性介质中进行氧还原反应(ORR)的经济而有效的电催化剂。例如,从聚苯胺和铁衍生的Fe-N-C催化剂被发现表现出ORR活性,其半波电位( E 1/2 )仅为 59 mV,比盈利的Pt/ C更低并且H2O2产率低于1%。最近,Feminus;Nminus;C催化剂中ORR的半波电位又提高了20minus;30 mV。此外,双金属(Fe,Mn)minus;Nminus;C催化剂已被证明具有改进的耐久性,在酸中进行了9000次电位循环后仅损失4%的活性。尽管取得了很大进展,但这些TMminus;Nminus;C催化剂与最先进的铂族金属(PGM)基催化剂之间仍存在相当大的性能差距。因此,必须对提高TMminus;Nminus;C催化剂催化性能的各种合成过程的机理有基本的了解。
研究表明,通过使用微孔炭黑作为载体、氨或氧蚀刻、或特殊载体方法来增加TM-N-C催化剂的微孔率可以显著提高催化剂的活性。对于ORR活性的增强,一个简单的解释可能是引入的孔隙扩大了暴露表面的总面积,从而增加了
TMminus;Nminus;C催化剂中可接近的活性位点的数量。然而,定量表征结果清楚地揭示了Fe-N-C催化剂对ORR的催化活性实际上与尺寸为5-15Aring;的微孔的特定面积相关,而不是暴露表面的总面积。因此,这一实验结果有力地表明,TM-N-C催化剂中的微孔可能具有催化ORR的高活性位点。受这个见解深入的实验结果的启发下,我们进行了第一原理密度泛函理论(DFT)的研究,研究了在微孔边缘形成的FeN4部分与在石墨烯层中形成的FeN4部分相比,如何增强ORR活性。
- 计算方法
在这项研究中,我们使用原子尺度材料模拟的计算机程序包(VASP)进行了自旋极化DFT计算。用投影缀加波(PAW)赝势来描述核心电子,用一个动能为400eV的平面波基来扩展波函数。在Perdew、Burke和Ernzernhof(PBE)泛函的广义梯度近似(GGA)框架内描述了电子交换和相关性。布里渊区使用Monkhorstminus;pack4times;4times;1 k点网格对活性部位FeN4minus;C10、3times;3times;1对FeN4minus;C12、4times;3times;1对FeN4minus;C8。在结构优化过程中,对原子位置进行优化,直到力低于0.02ev/Aring;。化学反应的过渡状态是用爬升图像推进弹性带(ClNEB)方法定位的,有6个中间图像,沿着和垂直于反应路径切线的力分量收敛为0.05eV/Aring;。本文报道的所有能量都包含了零点能量修正。ZPE校正计算为ZPE=sum;i 1/2hnu;i ,其中h普朗克常数,vi是结合分子的第一振动模式的频率。
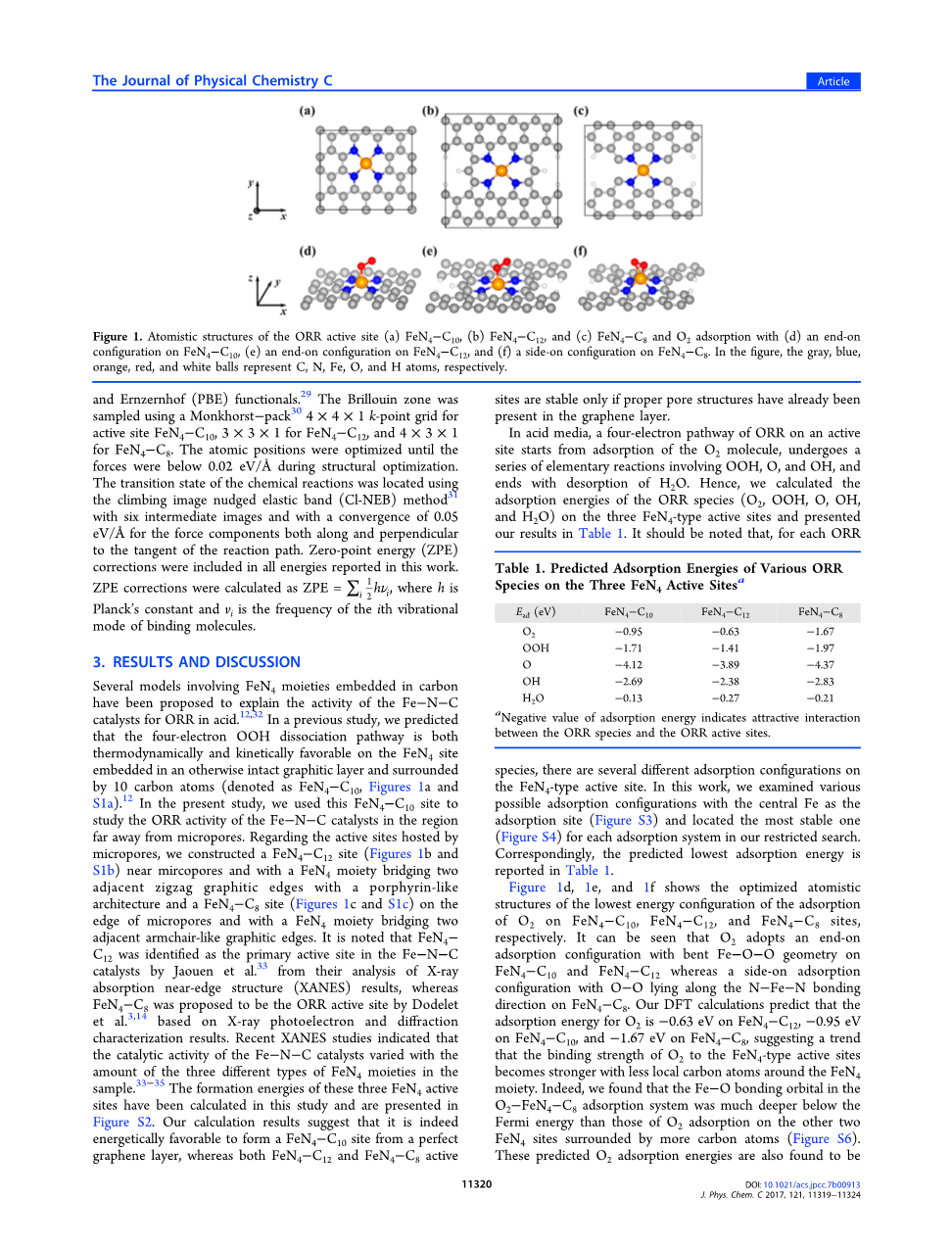
图1.ORR活性位点的原子结构(a)FeN4-C10,(b)FeN4-C12,(C)FeN4-C8和O2吸附,(d)FeN4-C10,(e)FeN4-C12端对配置,(f)FeN4-C8上的侧向配置。在图中,灰色、蓝色、橙色、红色和白色的球分别代表C、N、Fe、O和H原子
- 结果和讨论
提出了几种含FeN4基团的碳催化剂模型来解释FeN-N-C催化剂的酸催化活性。在之前的研究中, 我们预测四电子OOH解离途径在FeN4位嵌入在一个原本完整的石墨化层中的位置,并且被10个碳原子包围(表示为FeN4-C10,图1 a和 S1a)。在本研究中,我们使用FeN4-C10位来研究远离微孔区域的Fe-N-C催化剂的ORR活性。 关于微孔所承载的活性位点,我们在微孔附近构建了一个FeN4C12位点(图1 b和S1b),并且具有FeN4部分桥接两个相邻的锯齿形石墨边缘,具有卟啉样结构和FeN4C8位点( 图1 C和 S1c)边缘的微孔和分4部分桥接两个相邻的扶手椅状石墨边缘。值得注意的是,Jaouen等人通过对X射线吸收近边结构(XANES)结果的分析,确定FeN4C12为FeNC催化剂中的主要活性中心,而Dodelet等人基于X射线光电子和衍射表征结果提出FeN4C8为活性中心。最近的XANES研究表明,Fe-N-C催化剂的催化活性随样品中三种不同类型FeN4部分的量而变化。这三个FeN4活性位的形成能在本研究中计算并在图S2中呈现。我们的计算结果表明,它确实在能量上有利于从一个完美的石墨烯层形成FeN4-C10,而 FeN4-C12和FeN4-C8都是活性的,只有在适当的孔结构下,石墨烯层中的位置才是稳定的。
在酸性介质中,ORR在活性位上的四电子路径从O2分子的吸附开始,经过一系列的基元反应,包括OOH、O、OH、和 H2O的脱附。因此,我们计算了ORR种类(O2、OOH、O、OH和H2O)的吸附能对三个FeN4型活性位点进行了研究,并在表1中给出了我们的结果。值得注意的是,每个ORR在FeN4型活性位点上存在多种不同的吸附构型。在这项工作中,我们研究了以中心Fe为吸附位点的各种可能的吸附构型(图S3),并在我们的受限搜索中找到了每个吸附系统最稳定的一个(图S4)。相应地,预测的最低吸附能见表1。
表一、不同ORR三个FeN4活性位点上种类的能量预测
“吸附能负值表明ORR物种与ORR活性中心之间存在着吸引人的相互作用。
图1d、1e和1f分别显示了O2在FeN4-C10、FeN4-C12、FeN4C8位上吸附的最低能量配置的优化原子结构。可以看出,O2在FeN4-C10和FeN4-C12上采用端对端的吸附构型,Fe-O-O几何形状弯曲,而在FeN4-C8上采用侧对端的吸附构型,O-O沿着N-Fe-N键方向。我们的DFT计算预测O2在FeN4-C12、FeN4-C10、FeN4-C8上的吸附能分别为0.63 eV和1.67 eV,这表明O2与FeN4型活性中心的结合强度变强的趋势,FeN4周围的局部碳原子变少。事实上,我们发现O2-FeN4-C8吸附系统中的Fe-O 键轨道比O2吸附在其他两个FeN4位点上,被更多碳原子包围的位置(图S6)。这些预测的O2吸附能也被发现与预测的铂催化剂在Pt催化剂上(minus;0.69eV 在Pt(111)上和在Pt(100)上minus;1.10eV))的O2相类似。此外,我们在表1中的DFT结果表明H2O在FeN4型活性位上的结合比本体水的溶剂化稳定能弱约0.40eV。因此,我们在这里预测FeN4minus;C10,FeN4minus;C12和FeN4minus;C8活跃的位点能够吸引反应物O2引发ORR以及释放产物H2O以完成ORR。
遵循ORR的四电子途径,被吸附O2中的O-O键必须通过O2的直接解离或O2质子化后的OOH解离而被打破。这种O-O键断裂过程不受外部电极电位的影响。在表2中
我们报告了计算的三个FeN4型活性位点上两种类型的O-O键断裂反应(O2 vs OOH)的活化能。结果表明,在FeN4位上,O2直接解离反应的活化能总是高于OH解离反应的活化能。此外,发现O2加氢反应生成OOH的活化能非常低(例如,FeN4minus;C8上为0.04 eV)。因此,根据我们的DFT计算推断,对于FeN4型活性位点上的ORR,OOH解离途径在动力学上比O2解离途径更可行。
表2.FeN4活性位上O2解离和OOH解离活化能的计算
我们在表2中的计算结果表明,在FeN4-C8上的OOH离解反应只需要0.20ev的活化能,这比在其他两个FeN4型活性位上的活化能低至少0.36ev。此外,我们还发现FeN4-C8的中心Fe完全能够将OOH分子本身分解为O和OH(图2C, 图S5,表S2 )。相反,FeN4-C10和FeN4 -C12上的OOH分解反应预计需要将产物OH 运输到相邻碳的顶部(图2 a和2b)。因此,我们的DFT计算可以很好地预测OO 键的断裂在FeN4-C8上比在FeN4-C10和FeN4-C12位点上更容易,显然与在FeN4-C8上比在其他位点上更强的O2吸附有关。
图 2. 在(a)FeN4minus;C10上OOH解离反应的初始状态、过渡状态和最终状态的原子结构,(b)FeN4minus;c12和(C)FeN4minus;C8 活动地点。在图中,灰色、蓝色、黄色、红色和白色的球分别表示C、N、Fe、O和H原子。
除电位独立的Ominus;O键断裂反应外,ORR的四个电子途径包括几种质子化反应(如O2质子化、OH质子化和O质子化),其活化能随外电极电位的变化而变化。通过检测电极电位对ORR的影响,我们预测了OOH解离ORR途径对ORR的自由能演化三个FeN4类型的活动位点。这些自由能载点是使用表1中给出的吸附能和Noslash;rskov等人提出的计算方法(见 “ 自由能计算 ” 补充材料 )计算的。其中包括OOH分解为O和OH的步骤。非电化学反应的自由能变化和活化能与电极电位无关。因此,图3描述了相同的反应途径在其他研究中应用。我们的结果表明,所有的基本ORR反应都将是强烈的,因此因此在限制电极以下具有热力学优势,而一些涉及电荷转移的反应在该极限电极电势以上将变为负电荷。在图3我们绘制了在电极电位为0.69 V 下ORR的自由能演化图。
图3.计算了在酸性介质中,温度为300K,电极电位为0.69V时,活性中心FeN4-C10、FeN4-C12和FeN4-C8上OOH解离途径O2还原的自由能演化图。
此外,在0.69V下,最后一次OH质子化反应的自由能变化为零,但所有其他ORR步骤在FeN4minus;C10上仍进行的。因此,我们预测ORR在FeN4minus;C10上的热力学极限电位是0.69v。图3还表明,在0.69V以下O质子化反应需要在FeN4-C12上自由能增加 0.04 eV,而OH质子化反应在在FeN4minus;C8上的自由能增加0.15 eV。应该指出,我们对这三个FeN 4型活性位点的ORR极限势的预测接近于
剩余内容已隐藏,支付完成后下载完整资料
资料编号:[258080],资料为PDF文档或Word文档,PDF文档可免费转换为Word



