固液界面小分子吸附自由能的计算毕业论文
2020-06-17 21:31:01
摘 要
发生在固液界面的吸附现象是环境工程、化学工程、以及生物工程技术研究中的一项非常重要的内容。目前,在生命科学以及界面科学的研究中,生物分子在固液界面的吸附一直都是一个热点课题。众所周知,评价分子与材料作用的关键是计算出该分子在材料界面的吸附自由能,因此在界面吸附的相关研究中自由能的计算显得尤为重要。
羟基磷灰石(HAP)是一种具有高利用价值的无机物,是组成人体牙釉质和骨骼的最重要的成分,因此它通常被用来当做生物矿物的模型体系[1]。HAP的结晶、溶解和聚集过程通常和生命过程中很多复杂的现象有非常紧密的联系[2]。以生物矿化过程为例,在这一过程中液相的各种微观粒子想要进入矿物质中与其发生作用必须先通过HAP-水界面[3]。故我们选取HAP-水界面作为我们的一个研究对象。
石墨烯是一种新型的二维材料,自其被发现距今为止只有短短的十几年,却已被广泛应用于各个领域,这一切都归功于石墨烯自身的特殊结构。就目前的有关研究来看,石墨烯具有远超其它无机材料的导电性和导热性,是一种极具潜力的二维纳米材料。由于石墨烯这种独特优良的性能,所以对石墨烯界面的吸附进行研究有着重大意义。故我们选取石墨烯-水界面作为我们的另一个研究对象。
本文使用GROMACS4.5.5模拟软件,先采用分子动力学模拟(MD)的方法,通过伞状取样来计算固液界面小分子的吸附自由能,再对同样的体系用受控分子动力学模拟(SMD)的方法计算出另一组自由能数据与之对比。
关键词:固液界面 小分子 吸附 分子模拟
Calculation of Adsorption Free Energy of Small Molecule in Solid - liquid Interface
ABSTRACT
Solid-liquid interface adsorption is an important part of chemical engineering, environmental engineering and bioengineering. At present, in the life sciences and interfacial science, the adsorption of biomolecules at the solid-liquid interface has always been a hot topic. It is well known that the key to evaluate the role of molecules and materials is to calculate the adsorption energy of the molecule at the interface of the material, Therefore, the calculation of free energy in the study of interfacial adsorption is very important.
Hydroxyapatite (HAP) is a highly valuable inorganic substance that is the most important ingredient in the composition of human enamel and bones. It is often used as a model of biological minerals [1]. The crystallization, dissolution and aggregation processes of HAP are usually closely related to many complex phenomena in the life process [2]. In the process of bio-mineralization, for example, in the process of a variety of micro-particles in the liquid phase to enter the minerals and its role must first through the HAP-water interface [3]. So we selected HAP-water interface as one of our research objects.
Graphene is a new type of two-dimensional material, since its discovery has been only a short span of ten years, has been widely used in various fields, all thanks to the special structure of graphene itself. In view of the current research, graphene has far more conductive than other inorganic materials and thermal conductivity, is a great potential of two-dimensional nano-materials. Because of the unique and excellent properties of graphene, it is of great significance to study the adsorption behavior on graphene. We chose the graphene-water interface as another study object for us.
In this paper, GROMACS.4.5.5 simulation software was used to calculate the adsorption free energy of small molecules at solid-liquid interface by umbrella sampling and steered molecular dynamics simulation.
Key Words: Solid-liquid interface; Small molecular; Adsorption; Molecular simulation
目录
摘要 I
ABSTRACT II
第一章 绪论 1
1.1 引言 1
1.2 固液界面的吸附现象 1
1.3 羟基磷灰石简介 2
1.4 石墨烯简介 3
1.5 自由能的计算方法 3
1.6 选题的意义及研究内容 4
第二章 分子模拟 5
2.1 引言 5
2.2 分子模拟技术的发展现状 5
2.3 分子模拟方法 6
2.4 分子力场 6
2.5 边界条件 7
2.6 分子模拟的计算 7
2.7 GROMACS软件简介 8
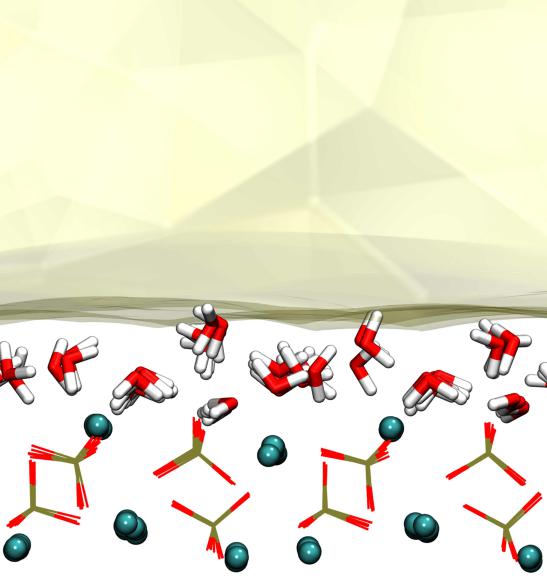
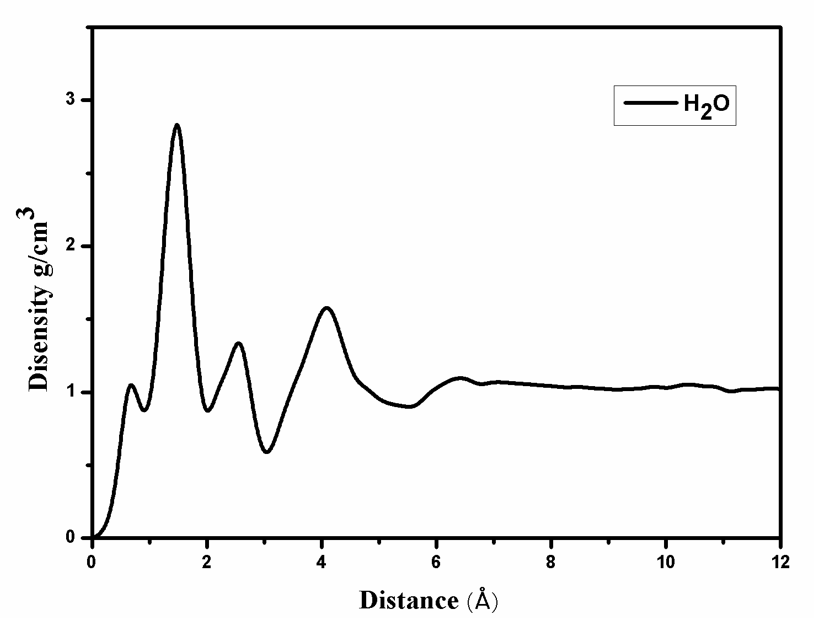
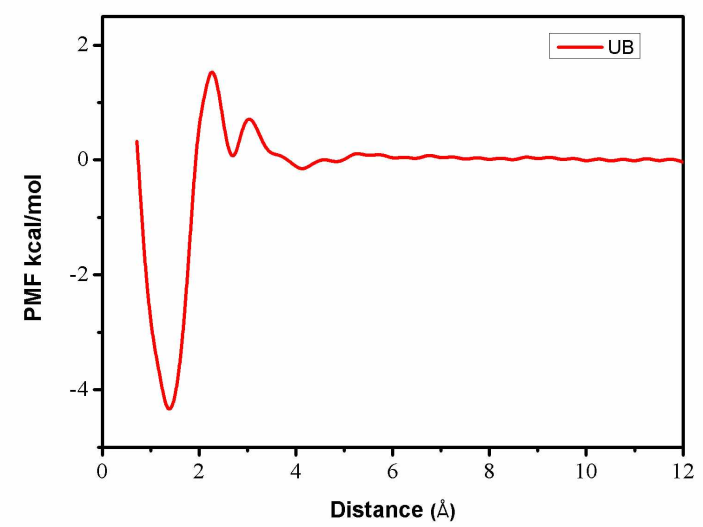
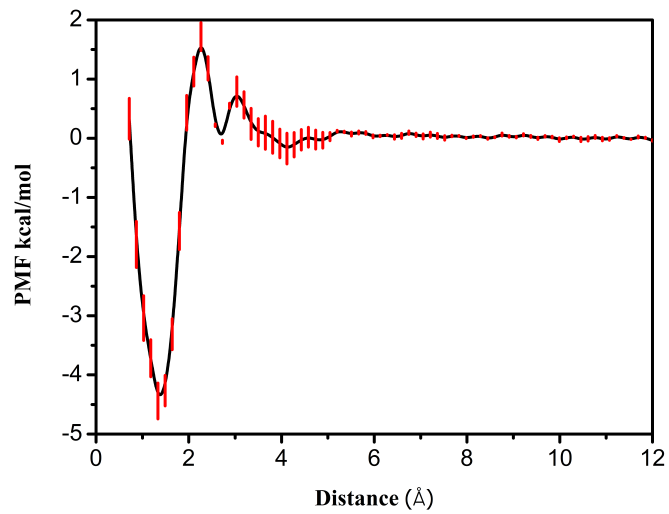
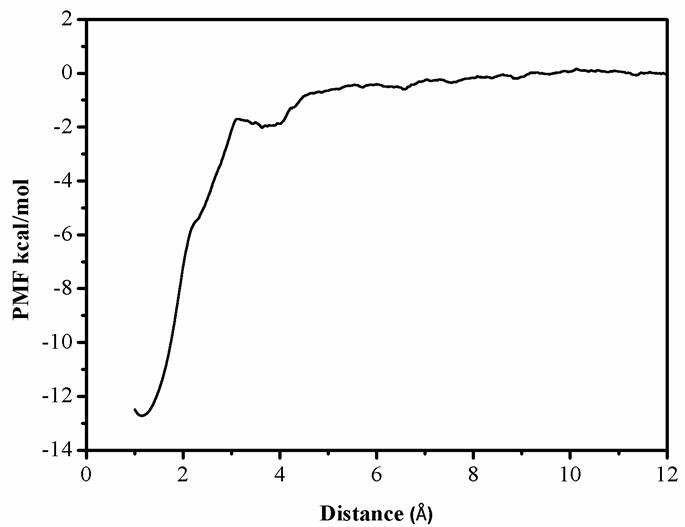
第三章 水分子在羟基磷灰石-水界面吸附自由能的计算 9
3.1 模拟方法 9
3.2 模拟细节 9
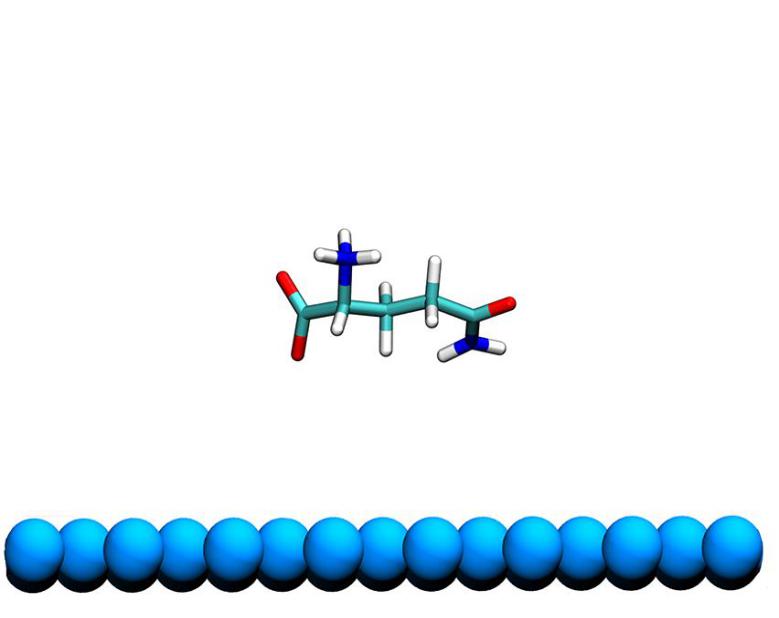
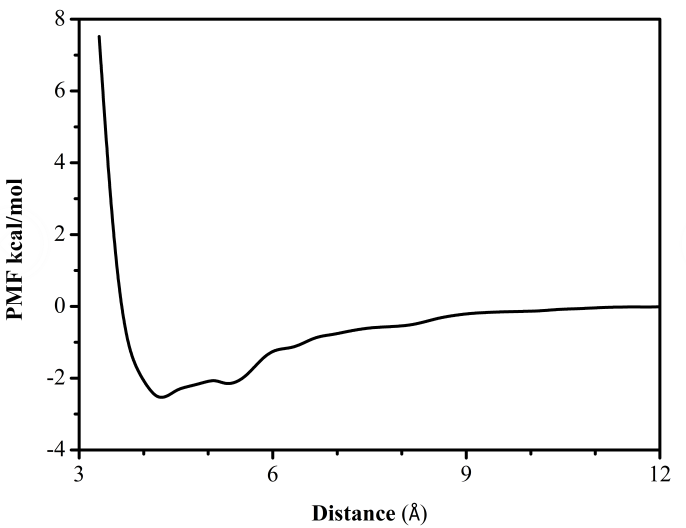
第四章 Gln分子在石墨烯-水界面吸附自由能的计算 14
4.1 模拟方法 14
4.2 模拟细节 14
第五章 结论与展望 17
5.1 结`论 17
5.2 展望 17
参考文献 18
致谢 20
第一章 绪论
1.1 引言
计算某一个过程的自由能一直都是分子模拟领域的难题之一,在计算自由能之前我们先要能够正确理解自由能的含义。这里我们以两个分子的结合过程为例,空间中的两个分子结合后,体系的总能量必然降低,在这个过程中释放出的能量就被称为结合能,两个分子结合之后引起了熵变,熵降低熵能却要增加,而自由能的变化就等于这一过程中的内能变化与熵能变化之和。自由能计算的方法多种多样,目前应用比较广泛的主要有伞状取样法[4],自由能微扰法[5-6],热力学积分法[7],Metadynamics以及Jarzynski Equality等。本文选取伞状采样和SMD模拟的方法作为自由能计算的主要方法,然而就伞状采样而言,在计算的过程中我们需要进行大量的样本采集和数据计算,受到研究体系和计算资源的很多限制,当把生物大分子作为研究对象时,计算会过于复杂,所以我们的本次研究只停留在小分子水平。本文主要研究水(H2O)和谷氨酰胺(Gln)等小分子的吸附自由能,这不仅仅是为了计算的便捷,同时也是为了能够给高分子(如蛋白质)在固液界面的吸附研究提供相应的理论和实验基础。
近年来在生物学和药学领域中产生了很多非常重要的研究方向,包括生物分子之间的相互作用[8],蛋白质折叠[9],离子通道[10]以及生物分子与材料的相互作用[11]等。对于上述的诸多研究方向而言,想要全面认识生物分子的结构与其功能,并且对生物材料做出正确评价。前提便是获取生物分子在材料界面处的吸附自由能。这也是我们课题研究的意义所在。
1.2 固液界面的吸附现象
发生在固液界面的吸附现象是环境工程、化学工程、以及生物工程技术研究中的一项非常重要的内容。自然界中,当固液两相发生接触,由于固体表面对液体分子的吸引力大于液体分子间本身的作用力,液相中的分子会向界面处发生移动并在界面上富集,这种现象就被称作为吸附现象[12]。研究固液界面的吸附作用具有很大的现实意义,因为它与人类的日常生活的方方面面都有着密不可分的联系。吸附作用在人类生活中的应用最早可以追溯到几千年前的布料印染技术。虽然吸附作用在生活和工业上的应用比比皆是,但由于固液界面,尤其是液相结构的复杂多样,导致目前还没有一整套独立完善的理论体系可以用来对固液界面的吸附做出详细的阐释。现在使用最多的是一种基于气体吸附理论的修正理论,其中添加的修正项多是从经验中推出,某些尚且没有充分的理论依据。近些年来,很多关于生物分子在固液界面吸附性质的研究课题已被相继提出,进行固液界面吸附的分子模拟研究对于攻克这些研究课题以及建立和完善相关的理论体系有着非常重大的意义。
1.3 羟基磷灰石简介
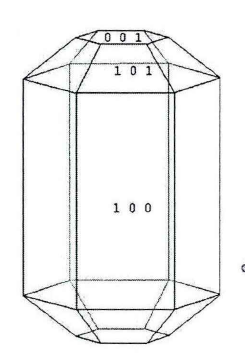 羟基磷灰石(HAP)是一种具有非常高的利用价值的天然矿物成分,其化学组成为Ca5(PO4)3(OH),它的晶体结构通常由两个分子组成,故分子式也常常写成Ca10(PO4)6(OH)2。自然界中存在的羟基磷灰石(HAP)主要有两种类型的晶相,一种是单斜P21/b型,另一种是六方P63/m型。而广泛存在于生物骨骼和牙齿中的HAP大多为六方晶型[13](如图1.1所示)。
羟基磷灰石(HAP)是一种具有非常高的利用价值的天然矿物成分,其化学组成为Ca5(PO4)3(OH),它的晶体结构通常由两个分子组成,故分子式也常常写成Ca10(PO4)6(OH)2。自然界中存在的羟基磷灰石(HAP)主要有两种类型的晶相,一种是单斜P21/b型,另一种是六方P63/m型。而广泛存在于生物骨骼和牙齿中的HAP大多为六方晶型[13](如图1.1所示)。
相关图片展示: