应变作用下多层MoS2光学性质的第一性原理研究毕业论文
2020-02-17 22:34:19
摘 要
二硫化钼是一种传统的润滑材料,但是在光电领域二硫化钼却是一种新型材料。单层二硫化钼作为一种新型二维半导体材料,因其优秀的半导体性质而备受关注。多层二硫化钼的性质大多介于单层和体材料之间。不同的是,多层二硫化钼的光敏感性十分优异,并且利用不同层数之间的差异可以制作不同波段的光电器件。但是不同层数之间的光学性质变化并不规律,在应用上有较大的难度。研究发现,多层二硫化钼的光学性质随应变会发生较为规律的变化。这一发现使多层二硫化钼光电器件的应用有了新的突破。
本文利用材料学分析软件MS(Materials Studio)并运用密度泛函理论进行应变作用下多层二硫化钼(MoS2)光学性质的第一性原理研究。文章详细论述了二硫化钼折射率,反射率,吸收光谱,消光系数等光学性质的推导方法和介绍了MS仿真的详细过程。本文重点分析了二硫化钼光学性质在应变下的变化趋势,以及应变作用对各个光学系数的影响。本文还在第一性原理分析结论的基础上,预测了应变作用下利用二硫化钼材料制作光学器件的可能。
关键词:多层二硫化钼;第一性原理;应变作用;光学性质
Abstract
Molybdenum disulfide is a traditional lubricating material, however it is a new type of material in the field of optoelectronics. Monolayer molybdenum disulfide, as a new type of two-dimensional semiconductor material, has attracted much attention due to its excellent semiconductor properties. The properties of multilayer molybdenum disulfide are mostly between monolayer and bulk materials. The difference is that multilayer molybdenum disulfide has excellent photosensitivity and can be used to fabricate photoelectric devices in different bands by using the difference between different layers. However, the optical properties of different layers vary irregularly and are difficult to apply. It is found that the optical properties of multilayer molybdenum disulfide change regularly with strain. This discovery has made a new breakthrough in the application of multilayer molybdenum disulfide optoelectronic devices.
In this paper, the first-principles study of optical properties of multi-layer molybdenum disulfide (MoS2) under strain is carried out by means of material analysis software MS (Materials Studio) and density functional theory. The derivation methods of optical properties of molybdenum disulfide, such as refractive index, reflectivity, absorption spectrum and extinction coefficient, and the detailed process of MS simulation are discussed in detail. In this paper, the variation trend of optical properties of molybdenum disulfide under strain and the effect of strain on optical coefficients are analyzed. On the basis of the first-principles analysis, the possibility of using molybdenum disulfide material to fabricate optical devices under strain is predicted.
Key words: multilayer molybdenum disulfide, first principles method, strain effect, optical properties
目 录
第1章 绪论 1
1.1 二硫化钼基本性质 1
1.2 国内外研究现状 1
1.3 研究目的与内容 2
第2章 基本原理与计算方法 4
2.1 密度泛函理论 4
2.2 第一性原理计算方法 5
2.3 二硫化钼光学性质计算方法 5
第3章 软件操作与参数设置 7
3.1 MS简介 7
3.1.1 CASTEP模块介绍 7
3.1.2 建模 7
3.2 参数设置 8
3.3 对应变的几点说明 9
第4章 结果与分析 10
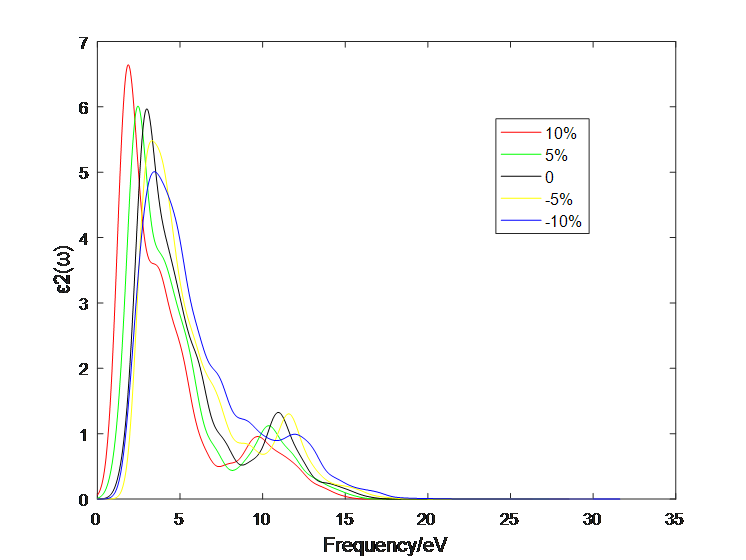
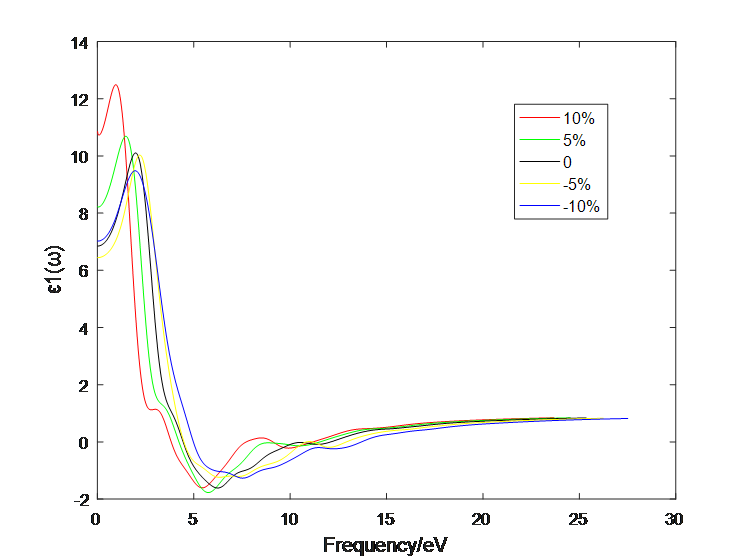
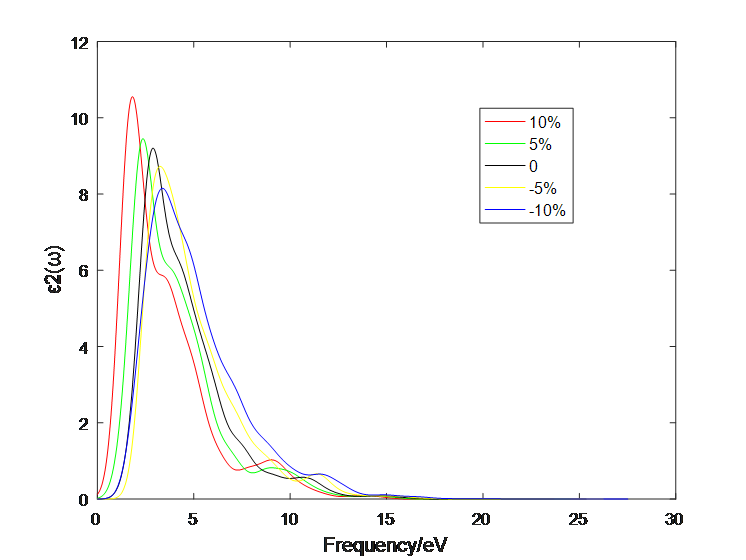
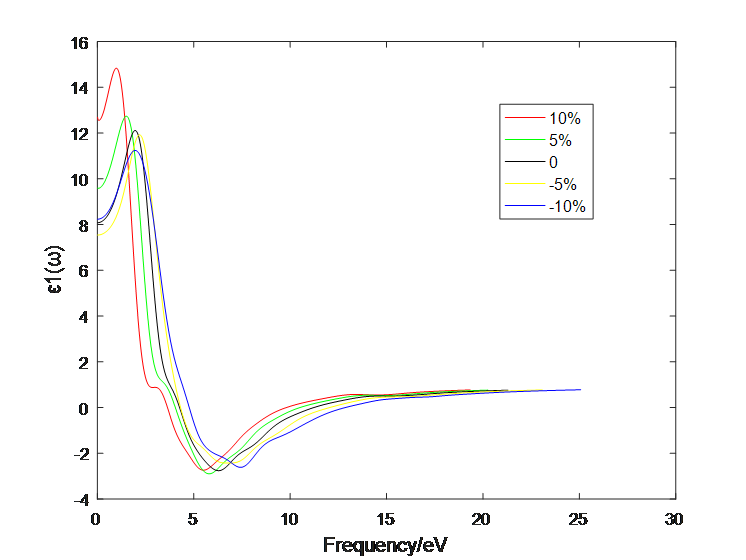
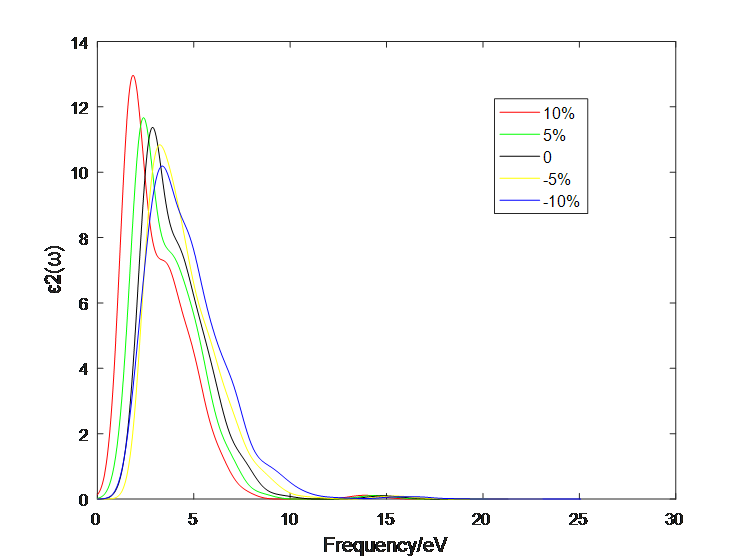
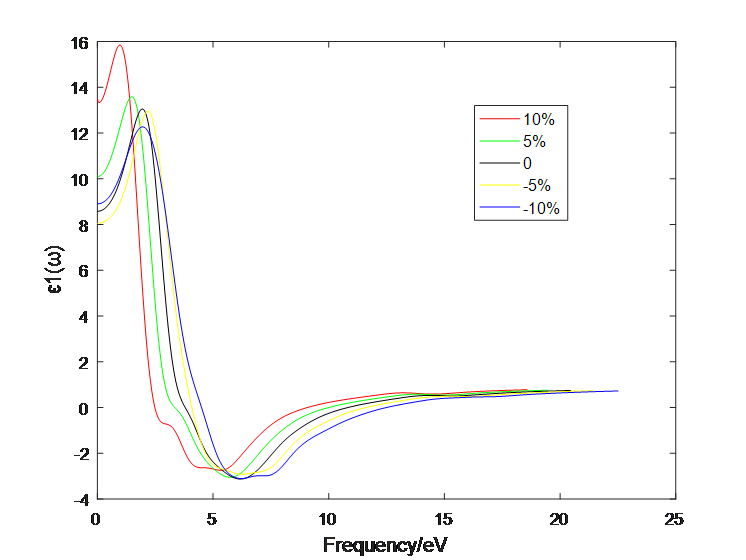
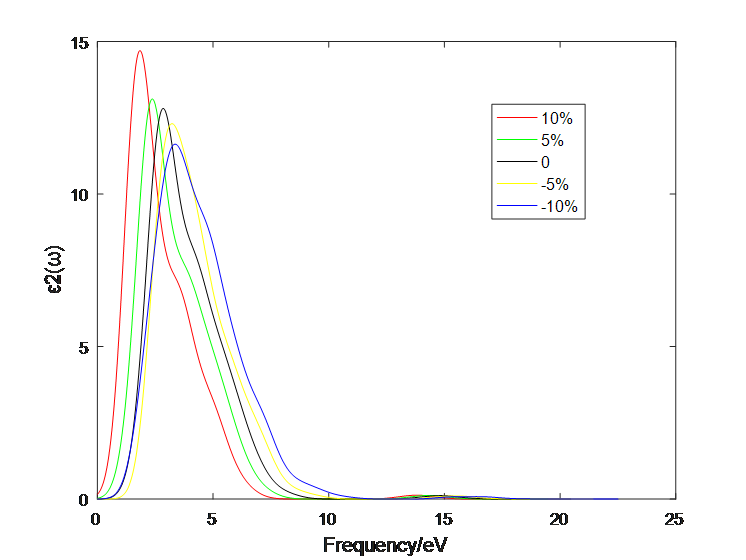
4.1 应变作用对二硫化钼介电函数的影响 10
4.2应变作用对二硫化钼光学性质的影响 13
4.2.1 单层二硫化钼 13
4.2.2 二层二硫化钼 16
4.2.3 三层二硫化钼 19
4.2.4 四层二硫化钼 22
4.3 本章小结 24
第5章 结论 25
参考文献 26
致谢 27
第1章 绪论
1.1 二硫化钼基本性质
二硫化钼是一种带隙不为零的过渡金属硫化物。二硫化钼的晶体是S-Mo-S三层原子组成的堆叠结构,其中硫离子与钼离子通过共价键连接。单层的二硫化钼是直接带隙半导体,它的禁带宽度约为1.8eV。晶体的二硫化钼具有和石墨烯类似的层状结构,层间通过范德华力连接,是禁带宽度约为1.29eV的间接带隙半导体。二硫化钼拥有石墨烯等材料所不具备的特性,比如很高的电子迁移率和发光效率。另外,二硫化钼具有稳定的化学性质和良好的热稳定性,表面活性高等优点。二硫化钼的上述特性使其在电子探针、固体润滑剂 、光电器件材料和离子电池正极材料应用等方面有着广泛的研究与应用。其中单层二硫化钼是一种新型二维半导体材料,具有轻薄透明的特性,在离子电池、气体探测器、发光二极管和新型晶体管材料等方向有着重要作用。多层二硫化钼材料的诸多性质介于晶体和单层的二硫化钼之间,其禁带宽度在1.2eV到1.8eV之间,由于其光敏感性良好,多层二硫化钼是一种十分理想的光电器件材料。
1.2 国内外研究现状
由于二硫化钼具有典型的层状结构,并且层间作用力较弱,容易被拆分,因此二硫化钼最早被当做固体润滑油使用,已有几百年的历史。直到最近制备工艺的日渐成熟,多层二硫化钼作为光电材料再一次进入了人们的视野。
多层二硫化钼的制备方法按是否发生化学反应可分为物理方法和化学方法[9]。物理方法包括机械研磨和超声剥离。化学方法制备二硫化钼的方法较为灵活,制备方法包括水热法、模板法、微乳液合成法、有机金属离子-剥离法、化学气相沉积法等。
在二硫化钼的光学性质方面,2014年,杨志鹏等运用密度泛函理论详细研究了二硫化钼的电子结构及光学性质[4]包括单层、多层和体材料。他们的研究结果表明,多层二硫化钼的光学性质随着层数的增减有一个明显变化的趋势,但是其变化并不规律,在实际应用中还不太具有可行性。他的研究结果为我们的研究提供了一些有价值的参考,比如,由于单层二硫化钼没有范德华力的影响,其电子结构和光学性质和多层二硫化钼相比呈现出较大的差异。
二硫化钼在应变作用下的光学性质方面的研究比较少,相关研究包括:2012年,吴木生,徐波实验组进行了应变对单层二硫化钼能带影响的第一性原理研究[5]。他们研究了双轴拉应变下单层二硫化钼电子结构的性质。其计算结果表明单层二硫化钼在很小的应变作用 (0.5%) 下,其能带结构就会由直接带隙转变为间接带隙。随着应变的增加,能带仍然保持间接带隙的特征,且禁带宽度呈现线性下降的趋势。同年,Emilio Scalise实验组发表了二维蜂窝结构的二硫化钼在应变诱导下存在从半导体到金属转变的报告[17]。实验发现单层和双层体系的带隙随着双轴应变的施加而减小。半导体-金属转变的拉伸应变约为8%,压缩应变约为15%。对于单层二硫化钼,在施加相对较小的应变(约2%)后,也观察到从直接带隙到间接带隙的转变。
在计算方法上,要计算二硫化钼的电子性质,最基本的方法是对构成该体系的薛定谔方程求解[1]。但由于二硫化钼材料体系中包含的粒子数量过于庞大,直接求解过于复杂,在现在的计算体系下还没有严格的解析解。故在实际的计算中,通常使用密度泛函的方法来简化计算。密度泛函理论的发展建立是一个很长的过程,是从一个又一个的简化近似方法中慢慢形成的。由Born和Oppenheimer在1927年提出的伯恩—奥本海默近似,其基本内容为:由于在质量上原子核比电子大很多,而在运动速度上电子却比原子核快许多,因此伯恩与奥本海默提出,可以近似的认为在某一时刻,考虑原子核运动时并不考虑电子的分布与影响,而考虑电子运动的时候将原子核近似为是静止不动的。这种计算方式被称为伯恩-奥本海默绝热近似[2]。同年,Thomas和Fermi提出的托马斯-费米模型为密度泛函理论建立了基础。1964年提出的霍恩伯格-科恩定理[14],充实了密度泛函理论的理论依据。在霍恩伯格-科恩定理的基础上,Kohn和Sham提出[15]了交换关联的方法,即用相互之间没有关联的粒子去替换体系中的粒子。在近似求解方法上,Kohn和Sham又提出局域密度近似法[16]。后来广义梯度近似又被提出来,与局域密度近似法形成互补。至此,在多粒子体系的密度泛函计算方法上趋于完整。
1.3 研究目的与内容
多层二硫化钼的物理性质特别是光学性质与层数密切相关。但是随着层数的变化,光学性质的变化并没有较好的规律,因此在实际应用上还需要更多的研究。如何改变并控制这种变化规律,使得多层二硫化钼表现出稳定变化的光学性质,是亟待解决的难题。目前,能较稳定影响二硫化钼光学性质的方法有:控制二硫化钼材料的分子层数、将二硫化钼材料与不同衬底结合[13]、改变二硫化钼材料工作的介电环境[7]、对二硫化钼材料施加应变作用等。比较上述方法,前两种方法在二硫化钼材料的制备工艺与衬底种类的选择方面还存在很大的难度,而改变介电环境在实际应用上显然不可行。于是,应变作用在这个情况下被提了出来。在已有的研究中,应变可以较为规律的改变二维结构二硫化钼的介电性质,而光学性质与介电性质又密切相关。于是可以推测,在应变作用下多层二硫化钼的光学性质变化是规律的。而通过应变作用影响二硫化钼材料的光学性质,有着操作简单,稳定,高效等优点。又考虑到目前对二硫化钼的研究主要集中在单层和电子结构,对于多层和光学性质的研究并不完善,特别是应变作用下的光学性质。因此,计算二硫化钼在应变作用下的光学性质变化规律对二硫化钼在光电材料方面的应用有着至关重要的作用。
本次论文的章节安排如下:第二章将会具体介绍本次研究中相关的基本理论与计算方法。第三章将会介绍本次研究中涉及的软件以及建模过程,参数设置和对应变作用的设定与说明。在第四章中,本文我们将对应变作用下多层二硫化钼的光学性质计算结果进行详细的分析。在第五章中,我们将会给出本次研究的结论。
第2章 基本原理与计算方法
2.1 密度泛函理论
密度泛函理论(Density Functional Theory)是一种用多粒子体系电子密度分布作为研究对象,进而推导体系基态性质的理论。Hohenberg-Kohn定理和Kohn-Sham方程奠定了密度泛函理论的基础,主要论述了处理多粒子系统的基态结构和性质的方法。
Hohenberg-Kohn定理的两个基本定理是:不计自旋的全同费米子系统的基态能量是粒子密度函数的唯一泛函;系统粒子数目不变时,通过电子密度分布对能量泛函取极小值,可以得到体系的基态能量。系统的能量泛函可以进一表示为:
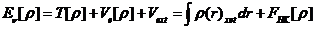 (2.1)
(2.1)
式中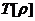 为无相互作用的粒子动能,
为无相互作用的粒子动能,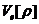 为库伦能,
为库伦能,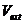 包括离子势和其它定域势,
包括离子势和其它定域势,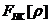 为体系交换关联能。
为体系交换关联能。
在Hohenberg-Kohn定理的基础上,Kohn和Shan更进一步提出了K-S方程。K-S方程的核心思想是:通过不相关粒子的电荷密度计算体系动能,影响总能量关键因素是库伦作用,而相互关联粒子的影响就要小得多。这时,可以用有相互作用的体系电荷密度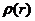 来表示单粒子轨道的贡献,即:
来表示单粒子轨道的贡献,即:
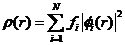 (2.2)
(2.2)
在这个情况下,体系的能量泛函可以表示为:
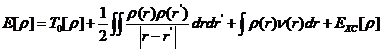 (2.3)
(2.3)
其中交换关联能:
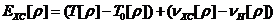 (2.4)
(2.4)
无相互作用的体系能量:
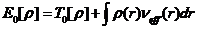 (2.5)
(2.5)
这时,无相互作用电子系统的有效能量:
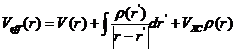 (2.6)
(2.6)
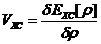 (2.7)
(2.7)
由上述式子可以得到K-S方程的表达式:
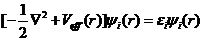 (2.8)
(2.8)
从而,体系的总能量泛函可以表示为:
 (2.9)
(2.9)
KS方程在此前的基础上减少了大量的计算,并且将体系基态的解通过基态密度分布表达了出来。
目前,能量泛函的近似方法主要有局域密度近似(LDA)和广义梯度近似(GGA)。其中局域密度近似是密度泛函近似的基础,其积分形式为:
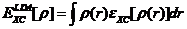 (2.10)
(2.10)
其中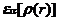 是相互关联粒子总能量,可以分为两个部分,即交换能和关联能:
是相互关联粒子总能量,可以分为两个部分,即交换能和关联能:
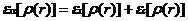 (2.11)
(2.11)
本次将会运用局域密度近似(LDA)进行第一性原理研究。
2.2 第一性原理计算方法
第一性原理计算获得体系基本性质的方式是利用量子力学的基本原理。第一性原理仅需要一些简单的物理参数(如电子质量、原子核质量等),不需要其他参数即可计算材料的物理性质和基态。赝势则是第一性原理能带结构计算中引入的一个虚拟的势,它有助于将多粒子体系复杂的计算进行近似简化。它的方法是将粒子体系形成的复杂的势替换为赝势,然后再进行总能量的计算。这些近似方法使得对材料物理性质的计算大大简化,在节约计算时间和简化计算条件的同时,计算精度也有一定的保障。随着理论的日渐成熟,第一性原理计算方法在材料研究中也得到了越来越多的运用。
2.3 二硫化钼光学性质计算方法
通过能带计算,可以得到二硫化钼的介电函数为一个虚函数。
介电函数公式[3]
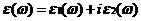 (2.12)
(2.12)
由二硫化钼介电函数得到光学性质的基础是复折射率与介电函数的关系,公式为:
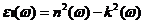 (2.13)
(2.13)
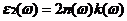 (2.14)
(2.14)
其中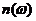 为折射率,
为折射率,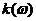 为消光系数。
为消光系数。
再由菲涅尔公式[10]可以得到反射率与折射率的关系:
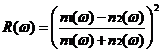 (2.15)
(2.15)
在得到折射率反射率等与复介电函数的关系后,可以进一步得到二硫化钼的几个光学性质与入射光频率的色散关系,包括固体的反射率、折射率、能量损失谱、吸收系数以及消光系数。公式如下:
以上是毕业论文大纲或资料介绍,该课题完整毕业论文、开题报告、任务书、程序设计、图纸设计等资料请添加微信获取,微信号:bysjorg。
相关图片展示: