MgO的晶格动力学与结构稳定性外文翻译资料
2021-12-25 16:41:17

英语原文共 9 页
MgO的晶格动力学与结构稳定性
摘要
利用密度泛函微扰理论,我们研究了MgO晶格动力学,电介质和热力学性质,NaCl-(“B1”)的P-T稳定场和CsCl-(“B2”)的结构相位。将结果与现有实验结果相比较,解决了MgO相图早期理论研究之间的争论。我们可以预测地幔条件B1结构是稳定的。静态计算预测B1-B2在490GPa时会发生转变;零点振动可将该压力降低16GPa。B2结构相位在110GPa以下是动态不稳定的,但在更高的压力下会变为动态稳定的。相反,B1相位在任何压力研究都不显示软模。在超高压下,MgO仍然是绝缘体:我们可以预测在20.7TPa下MgO 的B2结构相位的金属化。
介绍
氧化镁是一种对地球科学和固态物理学至关重要的材料:在地球上(尤其在地球的下地幔)它是最丰富的矿物之一,并且它也是一大类离子氧化物的原型材料。值得注意的是,氧化镁是已知的最小的多态性固体之一(仅有一个固相),并且在跨越压力高达227GPa和温度高达几千卡尔文的实验中NaCl-(“B1”)结构类型已经被发现。早期的理论计算发现,在向CsCl型结构过渡之前抗Nias型结构可能在高压下变得略微稳定,但后来排除了Nias和抗Nias结构,并提出氧化镁的NaCl结构向直接转化为CsCl结构相。事实上,对于高度离子型化合物,Nias和抗Nias结构都是被紧密的阳离子-阳离子或阴离子-阴离子接触破坏,它们的出现将是结合离子性降低甚至是金属化的迹象。尽管在足够高的压缩率下,所有的材料都应该变成金属,但预计氧化镁很难金属化。我们的计算表明,氧化镁的金属化发生在极高的压力下(20.7TPa)。
预测的B1-B2过渡压力较高,其最佳估计值为510 GPa。这超过了地球中心压力的40%。这种特殊的结构稳定性使得氧化镁适合作为高压实验的压力标准。根据原子间相互作用的精确表示,首次尝试创建基于氧化镁的压力标准已经在实验和分子动力学模拟中进行使用。
应该注意的是,在实验中还没有达到B1-B2的转变,从理论上讲,只有B1-B2在0 K时的转变压力是相对已知的(但不考虑零点振动)。最近的两项独立研究讨论了温度对B1-B2转变的影响,但得出了不同的结论。Strachan等人发现热效应相当温和。鉴于Drummond 和 Ackland的结果发现,随着温度的升高,过渡压力急剧降低,速率为每1000 K 50 GPa。
参考文献10中所用的方法是基于一个院子间势模型,该模型适合于静态从头计算伪势(原子电荷独立地被发现为10.7和20.7,氧化镁异常低值),而参考文献11是从头算冻结声子计算得到了对长程力的非常近似的描述。在构造动力矩阵和计算声子频率时,利用密度泛函微扰理论,可以获得更高的精度和更精确的长程力计算。这样的计算不仅能给出更精确的结果,而且能揭示以前使用的更近似方法可能存在的不足。Strachan等人还计算了氧化镁的熔化曲线,这是一个具有重要地球物理意义的课题,而且理论计算和实验之间存在重大的争议。本文所以的准谐近似法不能用于研究熔化,但可(参考文献10,另见参考文献16)通过检查相图上计算出的固体-固体(B1-B2)平衡线的准确性用于验证Strachan等人所用原子间势的有效性。
在下文中,我们给出了分离氧化镁多晶型稳定场的P-T平衡线的结果,以及在高压下这些相的一些性质(状态方程,热力学,介电性质,声子弥散曲线,声子密度状态和热力学函数)。我们的结果与现有的实验和理论研究非常一致,并且广泛支持了Strachan等人计算的氧化镁的相图(参开文献10)。
计算方法
目前的结果是通过使用Abinit代码获得的,Abinit代码是康宁公司鲁汶天主教大学的一个共同项目,也是其他贡献者基于伪电位和平面波得出的。它依赖于一种有效的快速傅里叶变换算法来实现实空间和倒易空间之间的波函数转换,依赖于对带共轭梯度法固定势的自适应,依赖于一种基于势的共轭梯度法来确定自洽势。关于计算对原子位移和均匀电场的响应的技术细节见参考文献14,随后的动力学矩阵,玻恩有效电荷,介电常数张量和原子间力常数的计算见参考文献15。计算是在曼彻斯特大学计算机中心CSAR的SGl Origin 3000 并行计算机上使用ABinit的并行版本进行的。
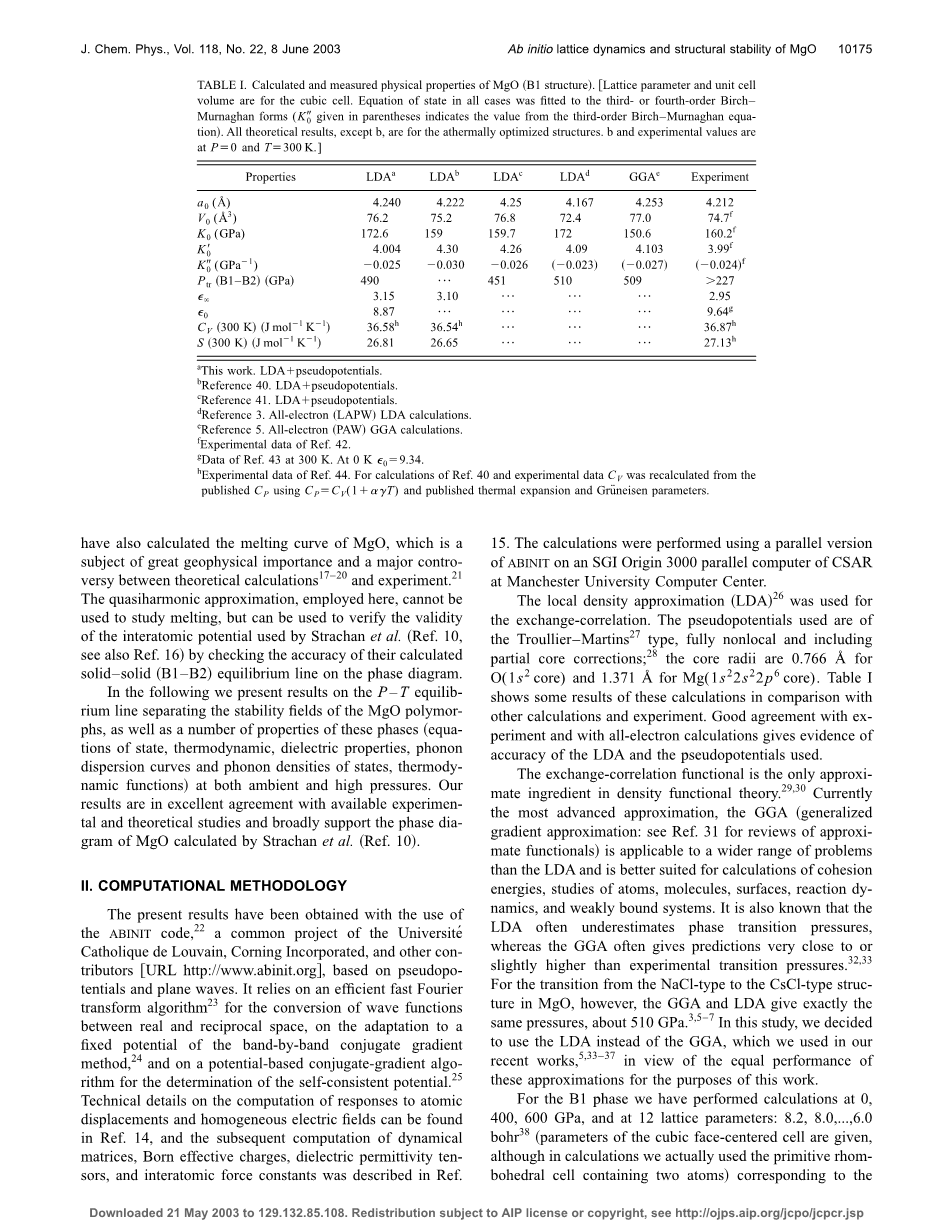
局部密度近似(LDA)用于交换相关。所用的伪电位为槽型-马丁斯型,完全非局部,包括部分芯校正;O的芯半径为0.766Aring;,Mg的芯半径为1.371Aring;。表一显示了这些计算的一些结果,并与其他计算和实验进行了比较。与实验和所有电子计算结果的良好一致性证明了LDA和所用伪电位准确性。
交换相关函数是密度泛函理论中唯一的近似成分。目前最先进的近似方法,GGA(广义梯度近似法:参见参考文献31了解近似功能的综述)适用于比LDA更广泛的问题,并且更适合计算内聚能,研究原子,分子,表面,反应动力学和弱束缚系统。众所周知,LDA经常低估相变压力,而GGA经常给出非常接近或略高于实验相变压力的预测。然而,对于从NaCl型过渡到CsCl型结构,GGA和LDA给出的压力完全相同,大约为510GPa。在这项研究中,我们决定使用LDA而不是我们在最近的作品中使用的GGA,因为在这项研究中,这些近似值得表现是相同的。
对于B1相,我们已经在0,400,600GPa和12个晶格参数下进行了计算:8.2,8.0,hellip;,6.0波尔(给出了立方面芯电池的参数,尽管在计算中我们实际使用了包含两个原子的原始菱形电池),对应于210到780GPa的压力范围。在0,400,600GPA和14个晶格参数下对B2相进行了计算:5.2,5.0,4.8,4.6,4.5,4.4,hellip;,3.6波尔,对应于216到1100 GPa之间的压力。
采用8*8*8的Monkhorst–pack网格对布里渊区进行采样,得出压力为0.01GPa,总能量为4* eV,多晶型之间的能量差为3* eV。平面波动能截止值为40 ha,压力收敛到0.04GPa,总能量为0.02 eV,能量差为4*eV。发现基态自洽(典型收敛eV).这些设置使声子频率收敛到2-5。
在包括G点在内的布里渊区的4*4*4 Q点网格上进行了响应函数计算,不仅提供了动态矩阵和声子频率,而且还提供了对电场,固有动态电荷和高频介电常数的响应。将Q点4*4*4网格上计算的动力学矩阵分解为短距离部分和长距离部分,对短距离部分进行傅里叶变换,得到与4*4*4实空间超单元(128个原子)相对应的短距离原子间力常数。我们核查了由于这个超级电池的有限尺寸造成的误差通常在2(对于极少数频率高达14)。从这些力常数加上长距离静电项(由玻恩电荷和介电常数描述),在稠密的Q点网格(通常为80*80*80或者更密集)上构造和对角化动力学矩阵,在每个体积上产生大量(几百万)个声子频率,用于计算状态g(w)的声子密度和热力学性质。
其中E0是静态晶格的能量。使用相同的力常数,动态电荷和高频介电常数,我们计算了声子色散曲线和静态介电常数。
结果
B1相的晶格动力学
在参考文献45和46中对氧化镁的声子色散曲线进行了实验研究;在参考文献43中对红外光谱进行了研究。Schu-tt等人的开创性计算证明了密度泛函微扰理论在晶格动力学研究中的巨大能力。随后,在参考文献40中用同样的方法研究了氧化镁的晶格动力学。参考文献48和11使用了对远程力进行修正的冻结声子技术。密度泛函微扰理论是一种较好的方法,因为它合理地,适当地考虑了长距离静电力,并且可以描述离子固体中的LO–TO分裂,且不需要对动态电荷和介电常数的值进行特别假设。
图1(a)表面理论声子色散曲线和实验声子色散曲线之间存在良好的一致性。如表一所示,我们的晶格动力学计算结果与实验结果非常吻合。图1页显示了理论声子色散400和600GPa时的状态曲线和声子密度(400GPa仍在B1结构的稳定场中,而600GPa则在B2结构的稳定场中)。在所有这些压力下,结构保持动态稳定,声子谱扩展到更高的频率,获得一个更复杂的结构,并且在频谱中间形成一个伪间隙。
B2相的晶格动力学
据我们所知,之前对该相晶格动力学的唯一研究是由Drummond和Ackland完成的。参考文献11图2显示了氧化镁的CsCl结构相在0,400和600GPa时的声子色散曲线,以及在400和600GPa时的声子态密度。这个相位是动态不稳定的,在0GPa下有整个软声子分支,如图2(a)所示。但是。它在110GPa以上(根据参考文献11,高于82GPa)动态稳定。动态不稳定性意味着,在非常高的压力下,热动力稳定的该相不能在110GPa以下减压。
介电常数和玻恩有效电荷
计算出的B1和B2相的玻恩动态电荷和介电常数如图3所示,是压力的函数。对于B2相,我们仅在其动态稳定性区域内显示这些性质。
在这两个阶段中,镁的动态电荷接近12的形式离子值,氧的动态电荷接近22的形式离子值,并随着压力的增加而缓慢下降。这证实了氧化镁的预期高电离度(在B2结构中更高)且其随压力的降低而降低,应该记住,动态电荷不是电离度的严格测量,例如参考文献49。
如表1所示,理论能够预测10%不确定度范围内的介电性能,众所周知,LDA高估了高频介电常数(讨论见参考文献50)。如图3(b)所示,两相的高频介电常数随压力变化不大,其初始值随压力有小幅度变化,随后,B1相的高频介电常数略高于320GPa,B2相的高频介电常数略高于900GPa。这可以通过使用克劳修斯-摩索蒂方程通过离子极化率来表示高频介电常数来合理化。
图1 .NaCl结构中MgO的声子色散曲线和声子谱(a)0GPa,(b)400GPa
,(c)600GPa
静态介电常数可以表示为高频常数(因为电子极化率)和离子位移极化率贡献的总和。考虑到的近似常数,静节点阐述随压力的快速下降主要是由于声子频率随压力的增加而引起的位移极化率的降低。
B1-B2相变
图4(a)显示了B1和B2结构及其交叉点的计算静态焓,对应于490GPa下的一阶相变,接近于所有电子计算中当前最准确的估计值510plusmn;5GPa。表二给出了通过将计算的E(V)数据拟合到Vinet得到两个相位的状态参数方程。
图3 (a)Mg的动态电荷与压力的关系图;(b)介电常数与压力的关系图
图4 MgO的B1-B2过渡(a)静态结果;(b)100K时的结果;(c)3000K时的结果
我们看到计算出的相平衡线是更类似于Strachan等人的研究结果,而不是德拉蒙德和阿克兰。当前结果与参考文献10中的结果之间的主要区别在于,它们的过渡压力比我们的低90GPa,比所有电子计算的低110GPa(这可能是由于它们使用的是伪电位),而斜率和我们的很近似。Drummond和Ackland在0 K(664GPa)下获得了更高的过渡压力,这显然是由于伪势误差所致的;它们的dP/dT斜率也有显著差异,可能是由于动态矩阵中的长程力和伪势误差的近似处理所致。
因此我们的结果总体上支持了Strachan的结果。对于固相,可以推测,他们使用的原子间势模型很可能产生接近B1和B2相熔化曲线的从头计算结果。在批评他们的方法之后,Strachan等人能够证明它们的熔化曲线是由两相分子动力学重现的。使用相同原子间势的模拟,因此最终只取决于该势模型的有效性。Strachan等人对于B1结构相的熔化曲线与现有的原子动力学和半经验分子动力学模拟结构一致。然而,所有的这些理论计算与实验结果之间存在严重的矛盾,实验结果显示高压熔化温度要低得多。
超高压金属化
根据我们的计算,在B1和B2的结构中,氧化镁在整个压力范围内(0-1000GPa)仍然是非金属。可以预测,氧化镁的金属化需要极高的压力:氧化镁是一种宽压缩隙的绝缘体,具有较低的压缩性,与Ne是等电子的,被认为是最难熔化的固体(金属化压力估计在158TPA和134TPA)。
我们使用两组计算非常密集的K点采样(24*24*24)和高斯涂抹技术(电子“温度”的0.2eV)探索了B2结构的金属化,一组计算采用LDA,在整个论文中使用相同的赝势以及更高的100Ha平面波截止。在这些计算中,发现金属化发生在1.95(九倍压缩,压力为13.4plusmn;0.2TPA)的单位晶胞体积。在这样的极端压缩赝势计算中与所有的电子计算相比,可以忽略较低的压力,因为忽略了芯-芯重叠。另一组计算使用的是GGA,冷冻芯电子爪方法在VASP62代码中实现,截止100毫米的平面波和200公顷的增强电荷。在这些计算中,我们发现金属化压力为20.7plusmn;0.5TPA。我们对金属化压力(优选值为20.7TPA)的估计证实了氧化镁是金属化最难的固体之一。
介绍
MgO是矿物物理和固态物理中最简单,最重要的材料之一。它是地球上最丰富的矿物之一,一种典型的离子氧化物,以及理论和实验方法研究最多的材料之一。在我们以前的工作中,使用从头算静态和分子动力学模拟,我们研究了它的状态方程,弹性,非谐性,以及在0 K时的结构稳定性。
在这项工作中。我们利用密度泛函微扰理论,研究了氧化镁从NaCl(“B1”)到CsCl(“B2”)结构的晶格动力学,介电性质和相变。这是密度泛函微扰理论在材料P-T相图预测中的首次应用(参见参考文献63和64)。这种方法具有良好的理论基础,并且结合高的数值精度与直接的固体热力学性质的方法,对于这样的应用来说肯定有很大的潜力。在这项工作中仅有的近似是:(1)LDA的交换相关能量;(2)赝势描述的核-价相互作用;(3)准调和近似。与所有的电子计算和实验比较表明,前两个近似(对于这里计算的性质)是适合的。准谐波近似是已知的分解温度超过50%-70%的熔融温度。在早前,使用从头算分子动力学模拟,我们已经表明,在准谐波近似中忽略的本征非谐效应对1 at
资料编号:[3735]



