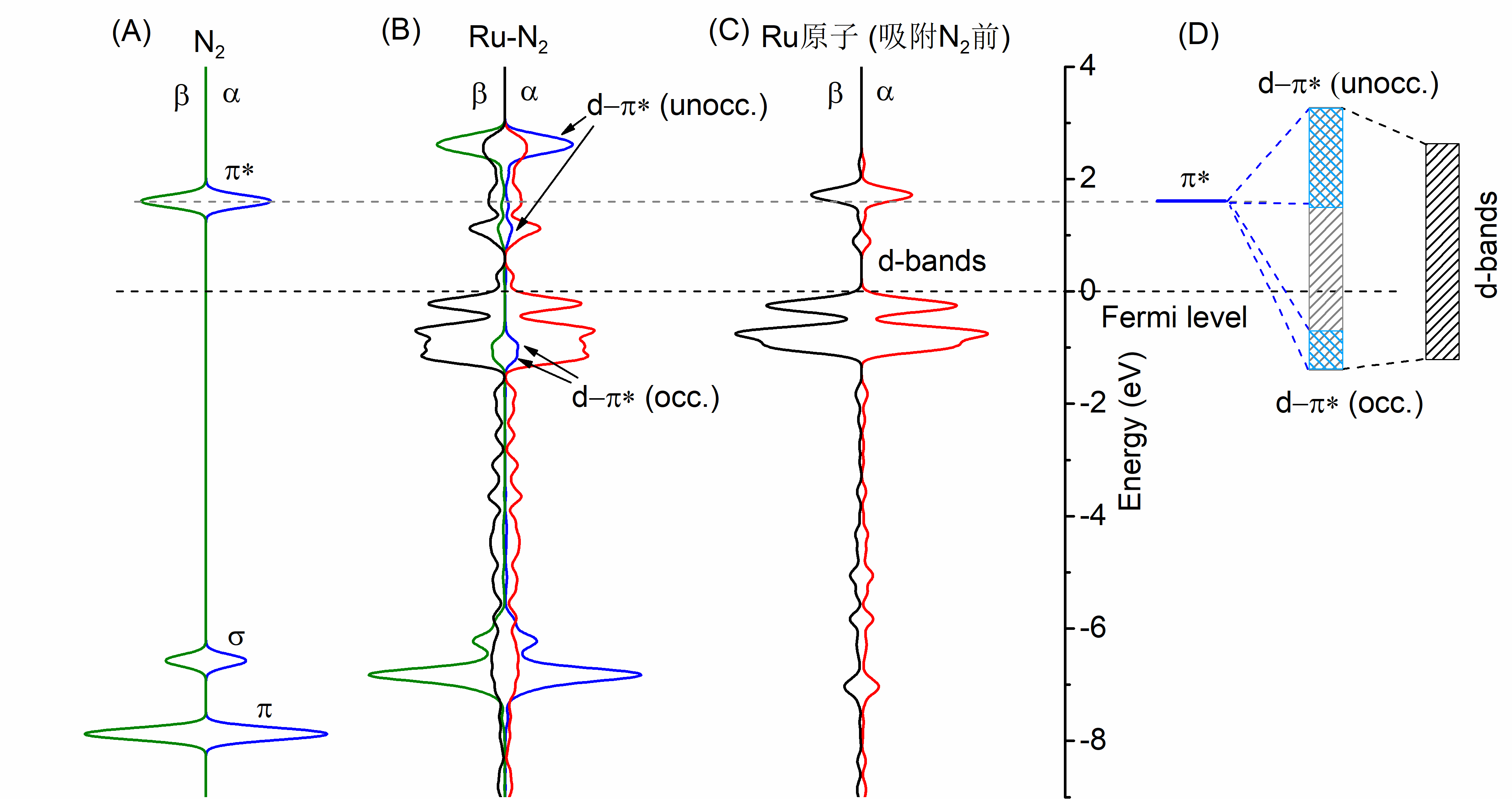
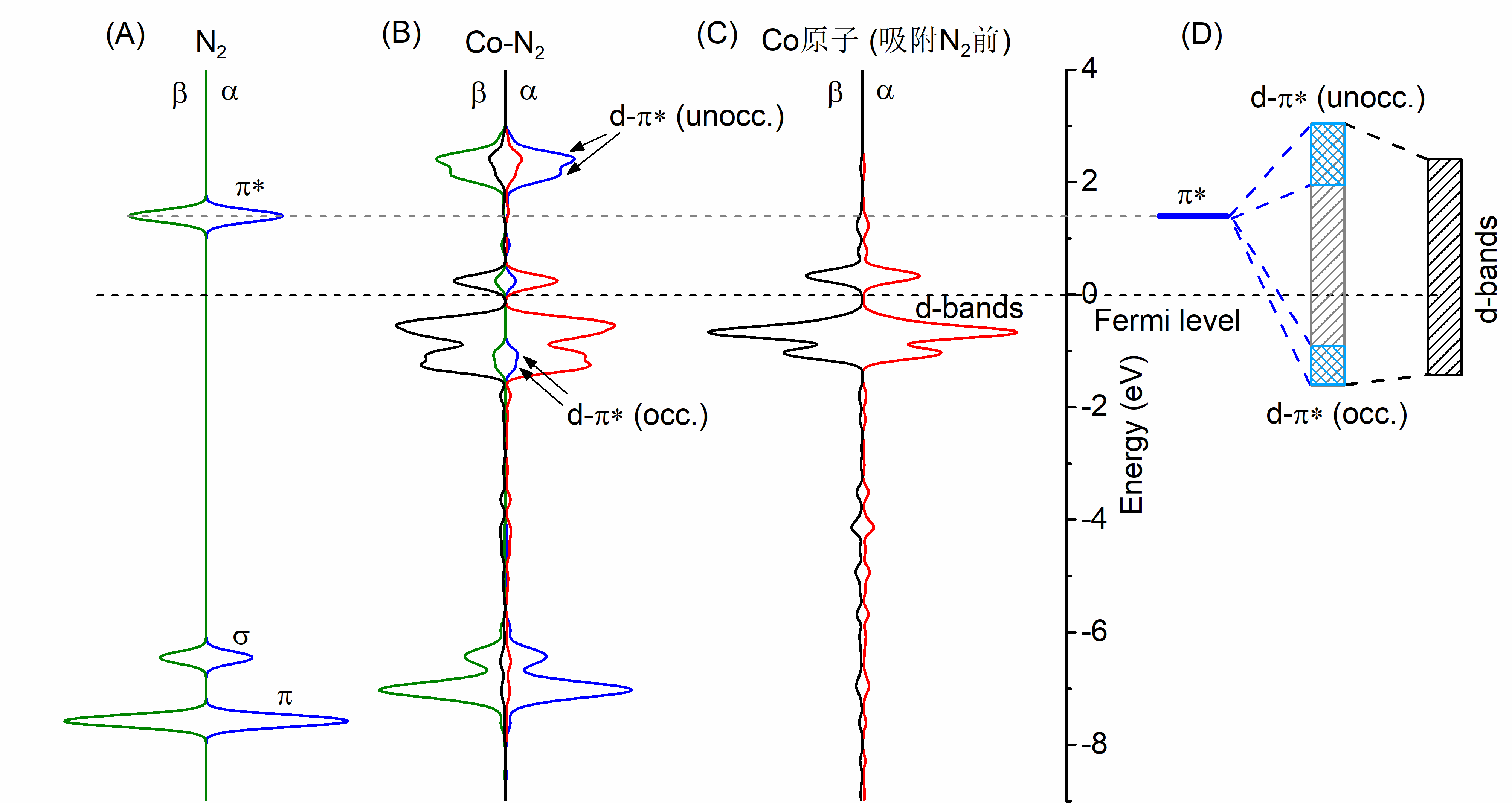
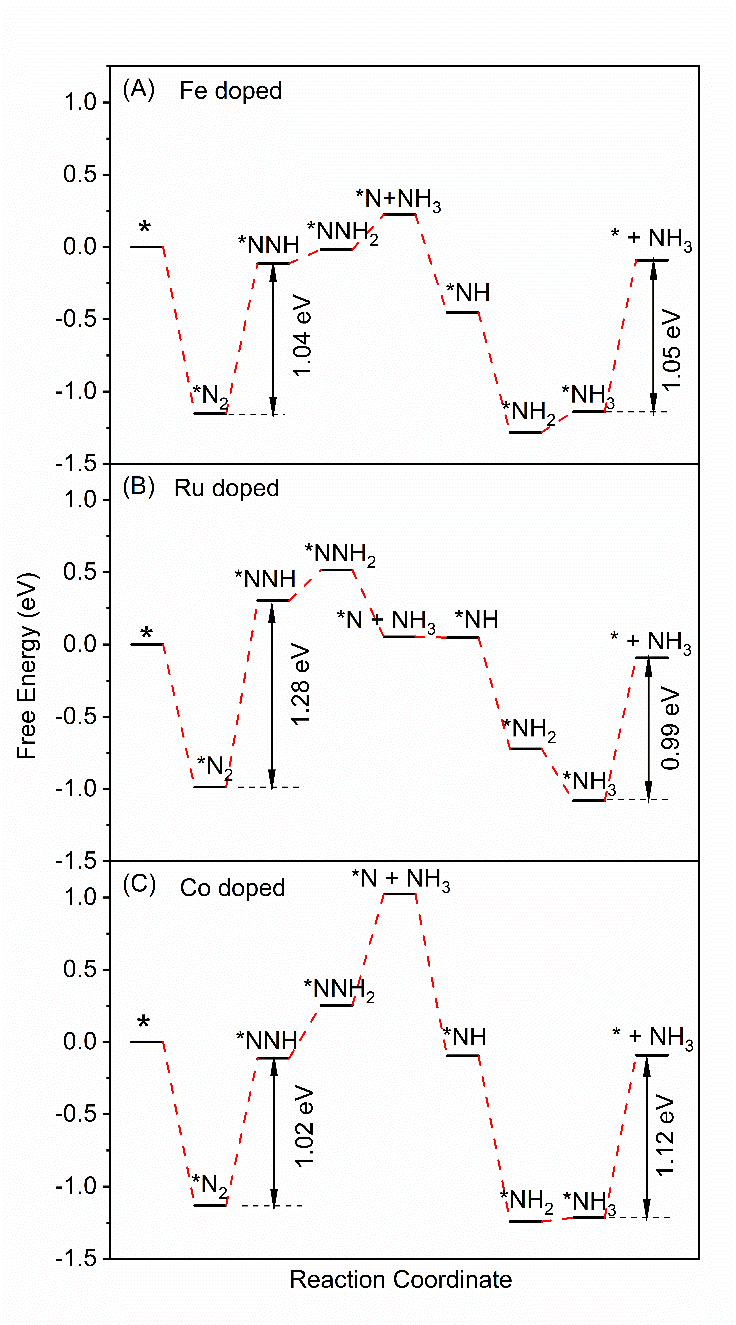
氮及金属原子共掺杂石墨烯电催化还原氮气的第一性原理研究毕业论文
2020-02-19 15:33:33
摘 要
氨气和铵盐是化学工业中生产最多的化学品之一。传统的工业合成氨主要采用的是哈勃-博施工艺,需要在中等高温及高压等较严苛的环境下进行,且消耗大量的能量,因此寻求在相对温和条件下的人工固氮技术一直以来是重要的研究方向。近年来新提出的单原子催化或单团簇催化是非均相催化中最前沿的研究领域。单原子催化剂具有确定的活性中心,为设计新的具有高稳定性、高催化活性以及高选择性的催化剂提供了新的可能。本课题以第一性原理计算作为研究手段,分别以三种过渡金属原子 (Fe,Ru和Co) 及氮共掺杂石墨烯为催化剂,研究并对比了上述三种过渡金属掺杂体系经几何优化后的电子结构性质以及氮还原反应机理。电子密度和差分电荷密度分析表明,在吸附氮气分子后,三种掺杂的过渡金属与氮气分子形成了化学键,此过程是化学吸附。分波态密度分析表明,三种掺杂的过渡金属的d能带分别与氮气分子的π*轨道发生了杂化耦合。另外,对各个反应物、中间体和产物进行结构优化,求得它们的Gibbs自由能,并绘制了它们的自由能阶梯图。对比三种体系的自由能阶梯图可知,氮气分子吸附到催化剂表面后第一步氢化还原和最后一步NH3脱附需要较大的能量,是整个反应的能量决速步。结果显示,三种体系中Fe掺杂体系拥有最佳的能量路径,反应决速步自由能变化为1.05 eV,相比之下催化性能最佳。而Ru掺杂体系在脱去氨气分子的时候拥有比其他两种体系更优异的脱附性能。
关键词:第一性原理;密度泛函理论;氮及过渡金属共掺杂石墨烯;电催化;氮还原
Abstract
Ammonia and ammonium salts are one of the most produced chemicals in the chemical industry. The traditional synthetic ammonia industry mainly adopts the Haber-Bosch process, which needs to be carried out under high temperature and pressure condition and consumes great amount of energies. Therefore, it is important to seek the artificial nitrogen fixation technology under relatively mild conditions. Single atom catalysis or single cluster catalysis, newly proposed in recent years, is one of the most cutting-edge research fields in heterogeneous catalysis. With explicit active sites, single atom catalysts provide new possibilities for designing new catalysts with high activity, selectivity and stability. In this study, the first-principles calculations were performed on three kinds of transition-metal (TMs) atoms (e.g., Fe, Ru and Co) and nitrogen co-doped graphene. The electronic structure properties and nitrogen reduction mechanism of these three systems were studied and compared after geometry optimizations. Electron density and charge density difference analysis show that when N2 adsorbing on three kinds of TMs doped systems, chemical bonds were formed between N2 and the doped TM, indicating the chemical adsorption process. Partial density of states (PDOS) analysis shows that the d-bands of three TM are hybridized with the π* orbital of N2, respectively. In addition, the Gibbs free energies of reactants, intermediates and products were obtained after geometry optimizations, and then plotted the free energy change diagrams of these systems. By comparing the free energy change diagrams of the three systems, it can be seen that the first step of hydrogenation reduction and the last step of NH3 desorption require large energy, indicating they are the rate-determining step of the whole reaction. The results show that the Fe-doped system has the energy favorable path among the three systems, with the Gibbs free energy change of 1.05 eV, which was the best performance among others, whereas the Ru-doped system has better desorption performance than the others when the NH3 is removed.
Key Words: First-Principles; Density Functional Theory; Nitrogen and Transition Metal Co-doped Graphene; Electrocatalysis; Nitrogen Reduction Reaction
目 录
第1章 绪论 1
1.1 研究背景 1
1.2 国内外研究现状 2
1.3 研究目的及意义 3
第2章 计算基本原理 5
2.1 第一性原理 5
2.2 密度泛函理论 5
2.3电催化氮气还原反应介绍 5
2.4 计算软件介绍 7
第3章 理论计算方法 8
3.1 计算参数设置 8
3.2 计算过程 8
第4章 计算结果与分析 12
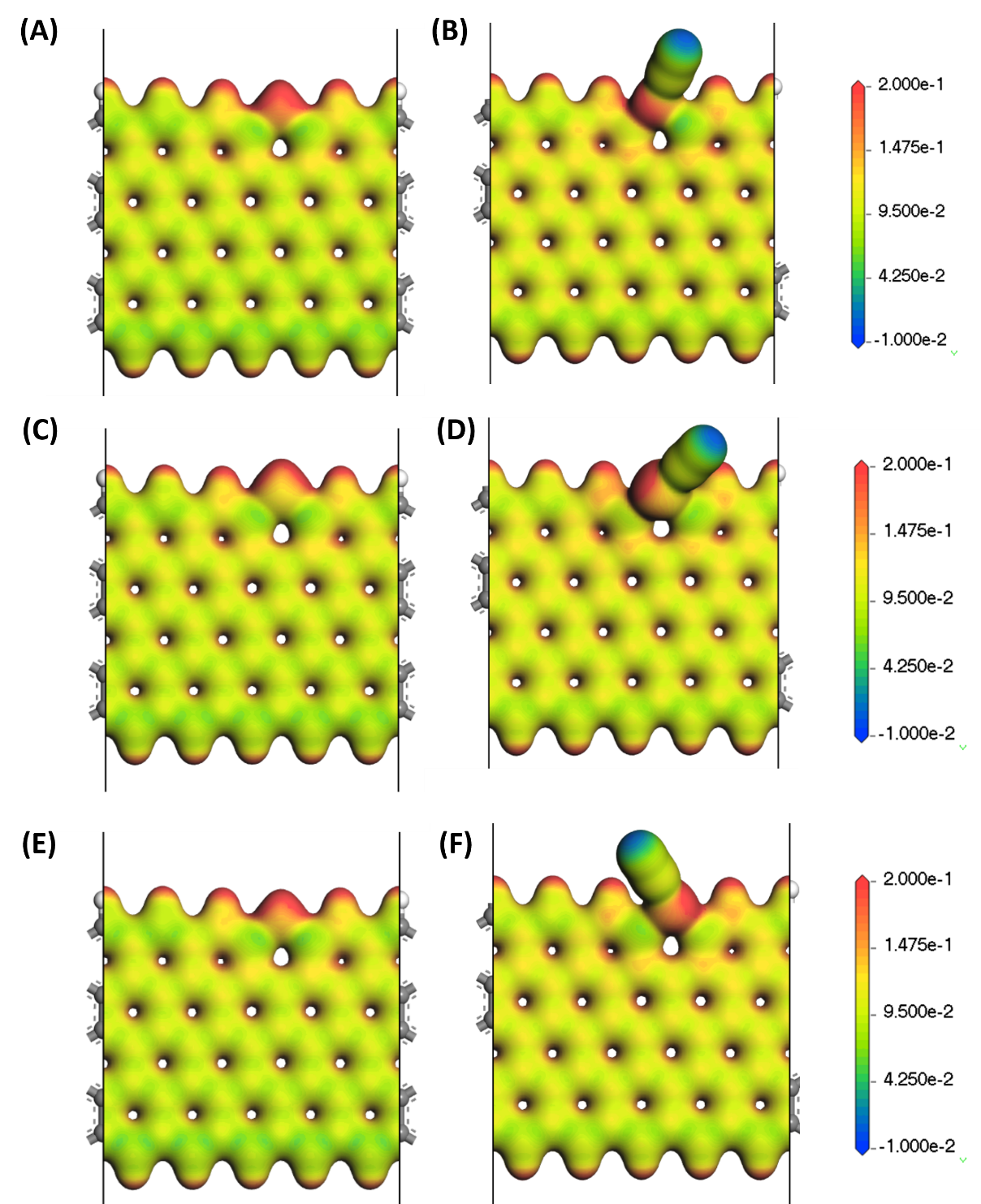
4.1 催化剂表面吸附氮气过程分析 12
4.1.1 几何结构变化 12
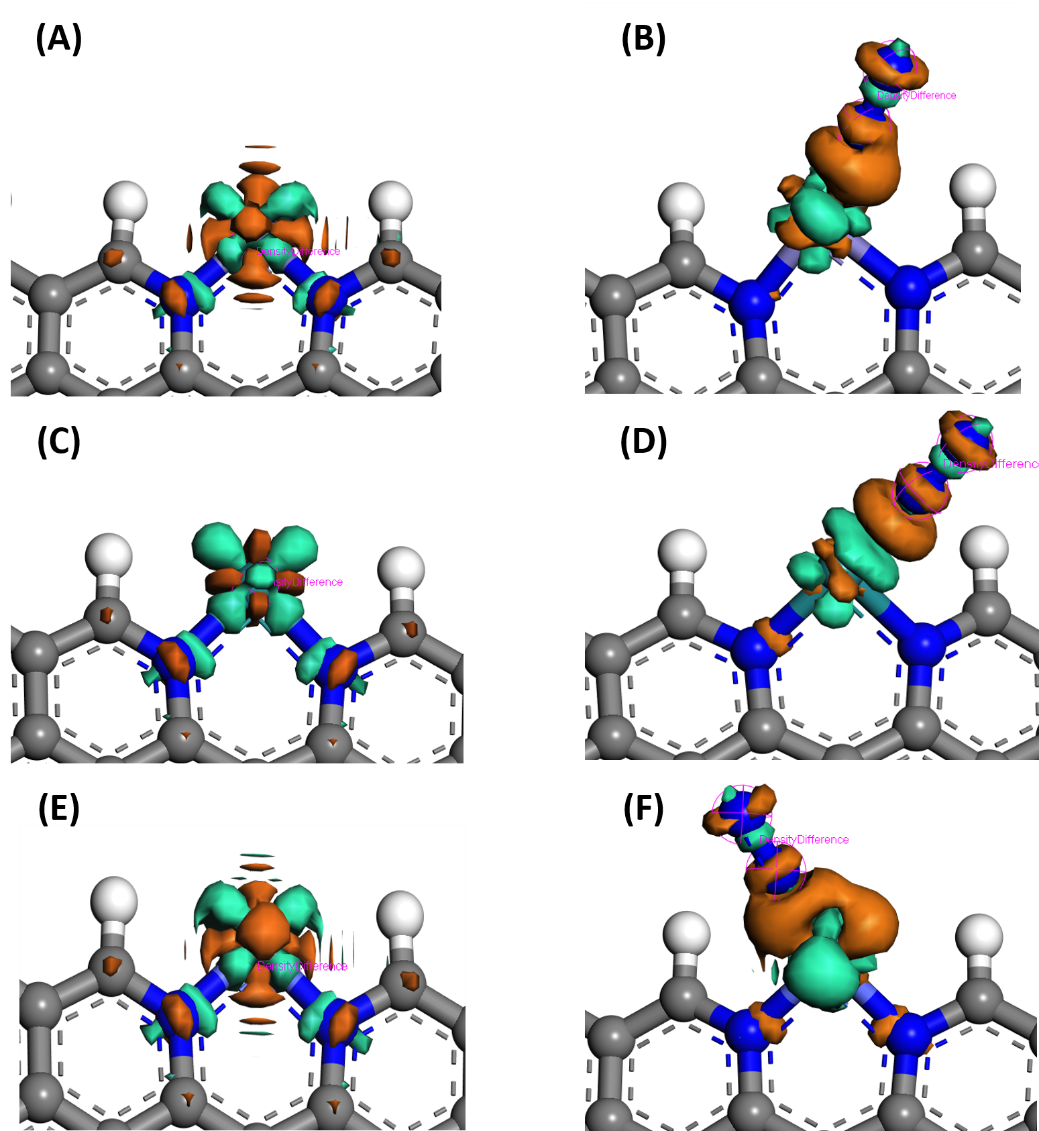
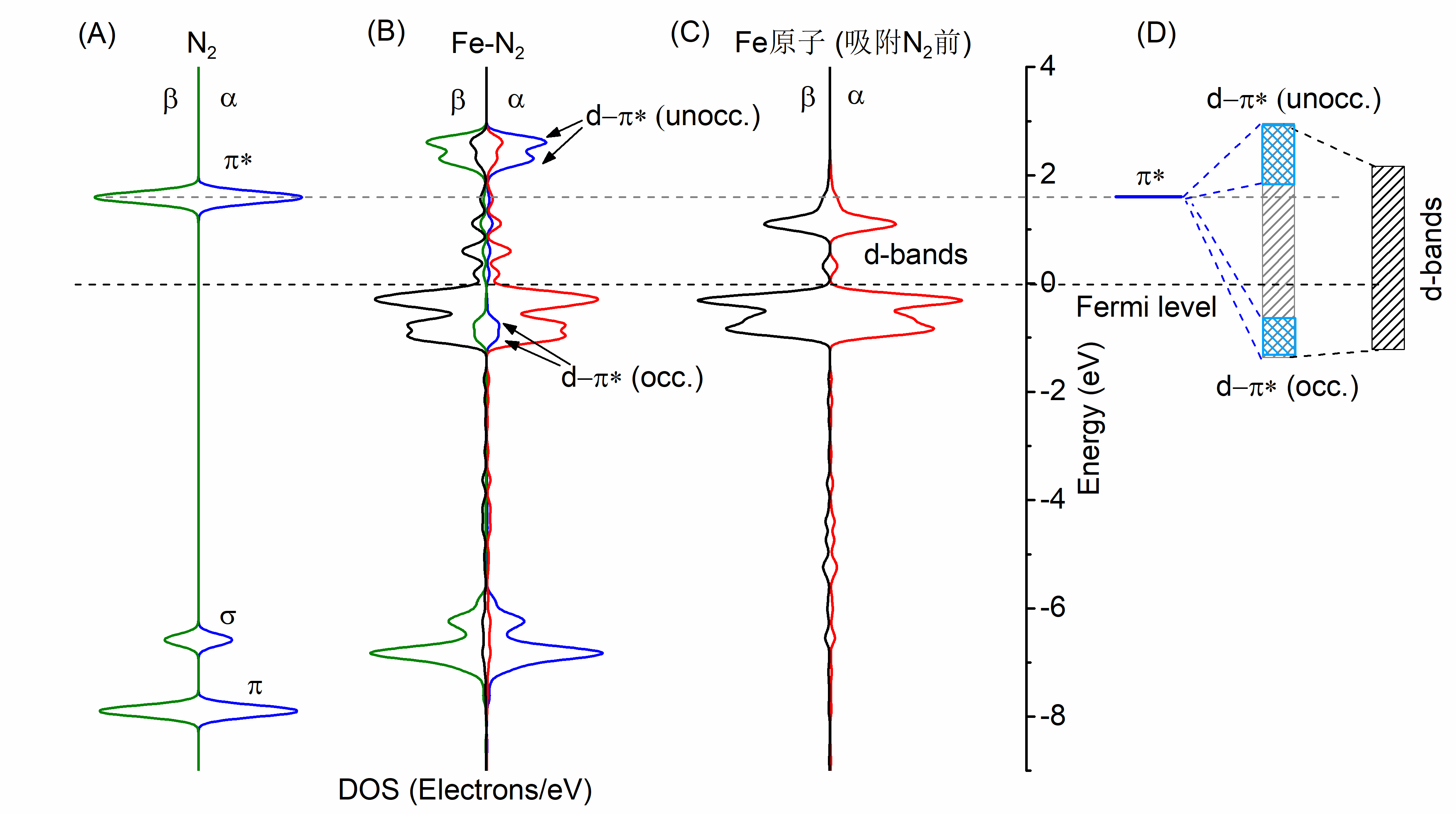
4.1.2 电子结构的变化和分析 13
4.1.3 吸附自由能分析 19
4.2 催化还原氮气性能分析 20
第5章 结论与展望 25
参考文献 27
致谢 30
第1章 绪论
1.1 研究背景
氨气和铵盐是化学工业中生产最多的化学品之一,其产量居于各种化工品的首位,是重要的化工、化肥原料。无论是加工合成用于农业的尿素、铵盐肥料,还是生产用于工业的染料、炸药、合成纤维、合成树脂,都需要大量的氨[1],可见其对于国民生产的重要经济意义。工业上生产氨气的原料为氮气与氢气。氮气资源丰富,约占大气成分的78%,然而还原氮气合成氨气并不容易。传统的合成氨技术主要采用的是哈勃-博施工艺 (Haber-Bosch Process,HBP)。这一工艺按照方程式N2(g) 3H2(g) → 2NH3(g) (ΔH 298°C = –92.4 kJ/mol) 进行。在哈勃—博施工艺中,N2和H2气体分子首先解离形成N和H原子,然后N和H原子反应形成化学键,生成产物NH3分子。而解离强N≡N三键需要很大的能量 (N≡N三键的键能为946 kJ/mol)。这一过程需要中等高温以及高压等较严苛的反应条件(约400 °C和150bar),并在铁基或钌基催化剂的作用下生产[2]。反应条件中的高温使反应物活化,提高了反应速率,但使得这一放热反应的反应平衡向左移动,高压条件使反应平衡向右移动,工业生产中需要综合考虑温度和气压两个条件,节能降耗,以提高生产的经济效益。由于这种高度能量密集的特性,合成氨消耗的能量占全球能量消耗的比例约为1—2%。此外,在工业氨合成过程中还会有大量天然气消耗与CO2排出,消耗天然气量占全球天然气总产量的2—5%,排放CO2量占全球CO2总排放量的约3%[3]。因此,寻求在相对温和条件下的人工固氮技术一直以来是重要的研究方向[4]。
在自然界的生物固氮过程中,氨可以通过称为固氮酶的酶系统合成,固氮酶可以由与豆科植物共生的根瘤菌等细菌合成,酶可在环境条件下以下列电化学反应产生氨:N2 8(H e–) → 2NH3 H2[5]。这些酶和电化学反应机理不同于工业上的哈勃-博施工艺。在生物电化学固氮的过程中,N2分子在酶促反应中依次被氢化,弱化N-N键,需要的能量比破坏N≡N三键更少 (断开N≡N三键中第一个键的键能为528 kJ/mol)。因此,寻求类似酶催化机理的催化工艺,从而替代哈勃-博施工艺是N2催化还原生产NH3的研究热点。
电化学催化氮气还原产氨作为一种新的合成氨方法,具有可在常温常压条件下进行、消耗的电能可以直接从可再生能源 (太阳能、风能等) 中转化获取等优点。国内外学者报道了大量电化学催化还原氮气的研究成果[5-6],然而目前的催化剂效果依旧不够理想,存在反应过电位较高、产物NH3选择性较低等问题。因此寻求更高效的催化剂、探究电催化还原氮气的反应机理是目前的研究热点[7]。
同时,随着计算机技术的不断革新,计算机技术与材料科学的交叉应用也成为了材料科学研究中的一种重要手段。计算材料科学的出现为计算机辅助进行材料理论改性或预测新材料的出现提供了可能。基于此,本课题拟用第一性原理计算为研究手段,以氮及过渡金属原子共掺杂石墨烯为催化剂,研究其对氮气电催化还原的性能及机理。
1.2 国内外研究现状
催化还原氮气产氨按照催化剂分类可以分为均相催化和非均相催化,目前工业上对非均相催化剂的研究包括单晶的铁催化剂及Al2O3、K2O等促进剂氧化物与非铁基催化剂。对单晶铁催化剂的研究表明,氨合成是一种结构敏感的反应,Fe (111) 晶面被证明具有高反应活性[8]。理论上反应活性来自高配位的Fe位点,它有最大的电子涨落。促进剂物质的存在极大地影响了HBP高温、高压环境中催化剂的结构和性能。在工业HBP催化剂中添加不会被还原的Al2O3、K2O等促进剂氧化物可防止铁的烧结,提高反应速率,并增加Brunauer-Emmett-Teller表面积。Ozaki等人[9]的研究提出催化剂表面上含氮物质的化学吸附和解吸的能量可能具有相关性,提出了吸附能“火山图”,有色金属催化剂的催化性能得以明确。铁、锇和钌处于“火山图”的顶部,表明这些元素的催化性能较好,这使得钌基催化剂成为了新的研究热点[10]。另外,用Al2O3、MgO、MgAl2O4和钡与铯的混合物促进剂的钌催化剂被证明对氨合成有效,从而产生了Kellog Brown and Root (KBR) 的商业化高级产氨工艺 (KAAP)[11]。KAAP于1992年在加拿大实施,其中Ba / Cs促进的Ru催化剂负载在石墨化碳上,其在低压下比HBP铁催化剂活性高十倍。Ru催化剂相比于Fe催化剂除了成本较高之外,还存在载体损失和催化剂寿命缩短的缺点。因此,对Ru催化剂和潜在的催化剂载体,如氮化硼,非热等离子体,BaCeO3纳米晶体,镧系元素氧化物 (CeO2,Sm2O3),石墨纳米线和沸石等的研究仍在继续。在最近的工作中,Hara等人[12]证明了基于氧化钙的电子([Ca24Al28O64]4 (e–)4) 作为Ru催化剂的电子给体,导致氨生成率相比于传统的氧化铝或氧化钙负载的Ru催化剂而言高达2120 μmol g–1 h–1 (进气口H2 / N2 (3:1),1个大气压,673 K),而前者氨生成率为50 至160 μmol g–1 h–1。
除了上述研究中的体相催化,近年来新提出的单原子催化(single-atom catalysis,SAC) 或单团簇催化 (single-cluster catalysis,SCC) 成为非均相催化中最前沿的研究领域[13-16]。单原子催化剂具有确定的活性中心,因此为设计新的高活性、高选择性、高稳定性的催化剂提供了新的可能。最近,Changhyeok Choi等人[2]通过密度泛函计算研究了固定在有缺陷的石墨烯衍生物上的单原子催化剂对氮的电化学还原反应 (NRR),发现与现有的块体金属表面上相比,由于在集团效应的帮助下SAC对析氢反应 (HER) 具有极大的抑制作用,SAC显著地改善了NRR的选择性。Li等人[17]应用第一性原理计算研究了θ-Al2O3 (010) 表面负载Fe3团簇作为非均相催化剂还原氮气合成氨,并发现其中吸附的N2首先被氢化成NNH的缔合机理主导了解离机理。此研究预测的合成氨的催化机理与工业上使用的Fe和Ru金属表面的催化机理明显不同。由此可见,电催化还原氮气产氨在反应机理的研究中仍有争论。
1.3 研究目的及意义
传统的催化剂研发受到研发成本高,实验周期长,试错代价大等因素的限制,开发新型高效的催化剂往往比较困难。随着计算机硬件的不断升级,运算能力的持续增强以及第一性原理方法研究的不断深入,应用第一性原理来研究催化机理,辅助催化剂的开发成为可能。采用理论计算的方法研究并筛选合适的催化剂,可以降低实验成本和缩短实验周期,并可以对催化机理进行原子层面的研究,对新型、高效催化剂的开发以及催化机理的研究都具有一定的指导意义。
基于上述因素,本课题以第一性原理密度泛函理论计算作为研究手段,以氮及过渡金属原子 (Fe, Co和Ru) 共掺杂石墨烯为催化剂,研究并讨论它们的电子结构性质,并以它们作为催化剂探索其对氮气电催化还原的机理;优化反应物、各中间体以及产物的几何构型,并计算它们的Gibbs自由能,寻找能量最优的电催化还原反应路径,期望阐明氮气电催化还原的微观反应机制;讨论和对比三种过渡金属与氮共掺杂石墨烯体系电子结构之间的区别,以及催化性能的差异。
第2章 计算基本原理
2.1 第一性原理
第一性原理计算 (First-principles calculation) 是一种完全基于量子力学理论的计算方法。与传统的化学,物理,材料等研究中通过实验观测、分析宏观现象来提炼内在规律获取经验模型方法或半经验模型的方法不同,第一性原理计算从构成物质的基本粒子,即原子核与电子出发,不使用经验参数,只采用电子质量、质子及中子质量、普朗克常量、电子电荷等基本的物理量,通过求解薛定谔方程 (Schrödinger equation) 得到优化的分子结构与分子能量,从而进一步计算物质的宏观物理性质[18]。第一性原理计算可以分为两大类,一类是基于波函数的从头算理论 (ab initio),另一类是基于电子密度泛函的密度泛函理论 (Density Functional Theory, DFT)。
2.2 密度泛函理论
密度泛函理论 (Density Functional Theory, DFT) 是一种基于量子力学理论研究多电子体系电子结构的理论方法。自二十世纪七十年代以来一直在材料计算中被广泛应用。该理论起源于Thomas-Fermi自由电子气模型,在Hohenberg-Kohn定理提出后得以完善而成为严谨的理论。H-K定理指出体系的基态能量是电子密度的唯一泛函,基态电子密度唯一决定了基态的波函数。1965年Walter Kohn和沈吕九进一步提出了Kohn-Sham方程,使得将电子密度作为基本变量来求解薛定谔方程具有可操作性。K-S方程成功地把求解含有3N个空间变量 (若将电子自旋考虑在内,则有4N个变量) 的波函数问题化简为求解三维空间的电子密度的问题[19],从而大幅度降低了计算复杂度。
2.3电催化氮气还原反应介绍
电催化氮气还原反应即通过电化学的方法使用催化剂还原氮气产生氨气或铵盐。在电解液环境里,质子来自溶液中的H3O ,外加电流提供e−,在催化剂的催化作用下,将催化活性位点吸附的氮气分子依次氢化,从而将氮气还原生成氨气或铵盐。对于催化剂催化氮气还原性能的影响因素,普遍重点关注的有催化剂的稳定性、催化剂对反应物及反应中间体的吸附与脱附性能、催化剂对氮还原催化的选择性、催化反应各步的反应过电位等。
Sabatier原理 (Sabatier Principle) 指出,催化剂与反应物及反应中间体之间的结合能既不能太大也不能过小。如果结合能太大,则反应物或反应中间体难以脱附,会导致后续反应持续进行,而无法获得预计的产物;如果结合能太小,则反应物或反应中间体难以与催化剂结合,会导致催化反应无法进行。因此,催化剂与反应物或反应中间体之间有合理的结合能是选择性催化的关键因素。在非均相催化剂上,氮气会首先结合在催化剂表面上,然后可能解离成两个吸附的氮原子,或者首先部分被还原成NNH然后继续解离成两个分子。根据Sabatier原理,最优的催化剂对关键反应中间体具有适当的结合能。在氮还原的体系中,关键中间体可能是NHx*或者NNHx* (这里x = 0–2,*代表催化剂表面)。
以上是毕业论文大纲或资料介绍,该课题完整毕业论文、开题报告、任务书、程序设计、图纸设计等资料请添加微信获取,微信号:bysjorg。
相关图片展示: